
Updated US FDA Guidance For The Gene Therapy Industry: Key Changes And Areas Of Increased Focus
By Leyla Diaz, Ph.D., Senior Technical Specialist, Biosafety Testing Services, Rockville, MD

In January 2020, the United States Food and Drug Administration (FDA) released several new or updated guidance documents pertaining to human gene therapy products. The guidance documents address product development and clinical trial design in general, as well as for specific disease indications. Two guidance documents that address product development, safety and characterization are:
- Testing of Retroviral Vector-Based Human Gene Therapy Products for Replication Competent Retrovirus During Product Manufacture and Patient Follow-up, which supersedes the guidance published in 2006
- Chemistry, Manufacturing and Control (CMC) Information for Human Gene Therapy Investigational New Drug Applications (INDs), which supersedes the guidance published in 2008
Several key changes that will impact developers of gene therapies are summarized below.
CMC Guidance Updates
The purpose of this FDA guidance is to inform sponsors of human gene therapy Investigational New Drug Applications (INDs), on how to provide sufficient CMC information required to assure product safety, quality and purity. The guidance applies to human gene therapy products and to combination products that contain a human gene therapy in combination with a drug or device. Among the key changes in the 2020 guidance are:
Reagents Used in Manufacturing
The updated 2020 guidance expands on the safety information requirement for reagents used in the manufacturing process. A particular focus is on reagents of animal origin that increase the risk of introducing adventitious agents in the final product. Materials of biological origin should be qualified with the appropriate documentation as to origin, manufacturing, and testing to ensure that they meet safety and quality standards appropriate for their intended use.
Monoclonal antibodies that directly contact the product during the manufacturing process should be characterized and tested for quality and safety as described in “Points to Consider in the Manufacturing and Testing of Monoclonal Antibody Products for Human Use”. This guidance provides recommendations on the characterization of cell lines used in the manufacture of monoclonal antibodies and product testing requirements for antibodies including those used as ancillary products. Careful consideration of these requirements should be given when selecting antibodies and recombinant proteins used in the manufacture of gene therapy products to avoid production and regulatory review delays resulting from insufficient reagent characterization.
Plasmids for Transient Transfection
The 2020 guidance also updates the recommendations for plasmids used as intermediates in the manufacturing of gene therapy products, in particular, AAV and lentiviral vectors. Plasmids should be derived from established bacterial master cell banks (MCB) that have been well characterized. Information on the plasmids should include manufacturing procedures and reagents. In addition, testing should include identity, sterility and endotoxin, levels of cell residuals (DNA, RNA, protein) and percent of supercoiled form. This information should be included in the IND submission regardless of whether the plasmids were made by the sponsor or a contract manufacturer.
Drug Substance Impurities
The updated guidance includes additional detail on characterization of product- and process-related impurities for the drug substance. Overall, it is recommended that the manufacturing process be optimized to minimize the level of impurities in the final product. Residual cellular DNA is highlighted as a common impurity with potential risk for biological activity that should be monitored and controlled in the final product. It is recommended that the size of the residual DNA be reduced to below 200 base pairs, well below the size of a functional gene, and that the amount of DNA be limited to 10 ng/dose. Furthermore, levels of transforming sequences, such as those present in HEK293 cells should be monitored in the final product with appropriate acceptance criteria to minimize patient exposure.
Additional impurities such as packaging of non-vector DNA and empty or non-infectious particles should be minimized, monitored and reported when applicable. Furthermore, residual reduction procedures and testing should be instituted for reagents used in manufacture such as cytokines, serum and growth factors, as well as antibodies and selection beads.
Replication Competent Retrovirus and Lentivirus Testing Guidance Updates
Because of the potential pathogenicity of replication competent retrovirus (RCR) and lentivirus (RCL), testing is required to exclude their presence in vector-based human gene therapy products. The 2020 guidance provides sponsors of human gene therapy products derived from the Retroviridae family of viruses with updated recommendations on testing for RCR/RCL during the manufacture of these gene therapy products and during follow-up monitoring of patients.
Table 1 summarizes the updated recommendations for product testing as compared to the previous guidelines.
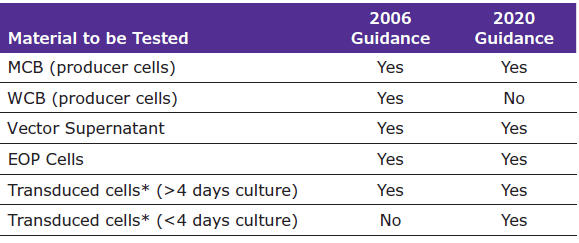
Table 1. Summary of updated recommendations for product testing.
*If data on a sufficient number of batches can be submitted to US FDA then this routine testing may be omitted.
Changes Pertaining to Cell Banks, Vectors and Drug Product
Retroviral Producer Cell Testing
Stably transfected vector producer cells have been genetically modified to produce retroviral or lentiviral vectors. The updated guidance state that both the cells and the supernatant from the MCB production should be tested for RCR/RCL. Testing of the WCB derived from an MCB that tested negative for RCR/RCL testing is no longer recommended.
Ex vivo Transduced Cells
When autologous or allogeneic cells are modified through retrovirus or lentivirus transduction, it is recommended that each lot of drug product be tested for RCR/RCL. The previous guidance stated that only ex vivo transduced cells cultured for longer than four days should be tested. The time exception has been removed in the updated guidance and the recommendation is now that all transduced cells, regardless of the time in culture, should be tested.
The updated guidelines also state that accumulated manufacturing and clinical data demonstrating that the ex vivo transduced cell product is consistently RCR/RCL-negative can be provided to the FDA to support the reduction or elimination of testing. If an agreement to reduce or eliminate testing is reached with the regulators, archiving sufficient cell product to perform RCR/RCL testing in the future is recommended.
Volume for Vector Supernatant Testing
The most significant change in the updated guidance is the recommendation regarding the volume of vector supernatant to be tested in the RCR/RCL assay. The 2006 guidance recommended testing 5% of the total production volume, to a maximum of 300 mL, for production volumes at or above 6 L.
The updated guidance still recommends that, in all cases, at least 5% of the total supernatant should be tested by amplification on a permissive cell line. However, it is also recognized that this recommendation may not be appropriate for production volumes currently used in clinical manufacturing. For volumes larger than 6 L, it is recommended that enough supernatant be tested to ensure 95% probability of detecting one RCR per dose. Calculating the volume of supernatant to be tested independent of lot size involves determining a dose equivalent using direct administration or cell transduction parameters. The guidance provides instructions for calculating testing volumes based on the Sponsor's specific dosing regimen. It should be emphasized that knowing vector transducing units is necessary to calculate testing volumes; this should be considered when establishing production sampling plans to collect material for RCR/RCL testing.
Summary
As our collective knowledge of gene therapies and experience in their manufacturing continues to increase, guidance from regulatory agencies will be critical to continued growth of the industry. Risk mitigation strategies defined by the FDA and other agencies will evolve in order to ensure product quality and patient safety. While much of the finalized 2020 guidance is consistent with earlier draft guidance, key changes emphasize the importance of working with a partner with regulatory expertise in this field to help develop and execute the optimal gene therapy testing program.