
Understanding the Three Major Challenges Limiting Innovation In Tissue Engineering
By Lev Gerlovin, Andrew Thomson, and Jack Vailas, Life Sciences Practice, CRA
The regenerative medicine sector, comprised of tissue engineered products and cell and gene therapies, has grown rapidly over the past decade, including significant therapeutic advances that offer the prospect of optimizing and transforming patient care. There has been a dramatic increase in the number of approved cell and gene therapies and available funding for these products, and yet both progress in development programs and approvals for tissue engineered medical products (TEMPs) have lagged behind (see Figure 1). While there are currently more than 1,000 companies developing cell and gene therapies, only about 130 of them are exploring TEMPs as a viable source of regenerative therapeutics. This delayed maturation of TEMPs is largely due to the added complexity that these products present in terms of their structural composition and manufacturing processes compared to cell and gene therapies. Our Life Sciences Practice team at CRA recently conducted an analysis of product development programs in tissue engineering and found that there are three key hurdles restraining the growth of this sector:
rapidly over the past decade, including significant therapeutic advances that offer the prospect of optimizing and transforming patient care. There has been a dramatic increase in the number of approved cell and gene therapies and available funding for these products, and yet both progress in development programs and approvals for tissue engineered medical products (TEMPs) have lagged behind (see Figure 1). While there are currently more than 1,000 companies developing cell and gene therapies, only about 130 of them are exploring TEMPs as a viable source of regenerative therapeutics. This delayed maturation of TEMPs is largely due to the added complexity that these products present in terms of their structural composition and manufacturing processes compared to cell and gene therapies. Our Life Sciences Practice team at CRA recently conducted an analysis of product development programs in tissue engineering and found that there are three key hurdles restraining the growth of this sector:
- Complex manufacturing requirements relative to those for cell and gene therapies that present significant challenges for scaling production and quality control;
- Undefined approval pathways leading to inconsistencies in the time, cost, and data required to bring TEMPs to market; and,
- Insufficient reimbursement streams and a lack of innovative funding options.
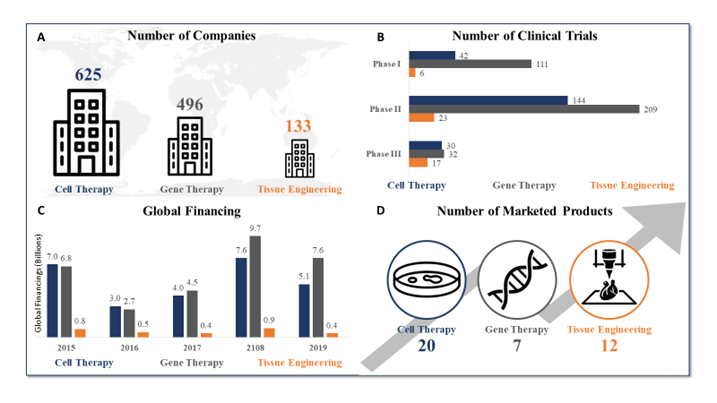
Figure 1: Temporal comparison of cell therapy, gene therapy, and tissue engineering (Source: Data from the Alliance for Regenerative Medicine, July 2020)
Complex Manufacturing Processes
The manufacturing process for TEMPs is especially challenging for many drug developers, more so than for cell and gene therapies, due to unique complexities including the required raw materials, uncertainties regarding quality control, and issues in scaling production capabilities. This has deterred some companies from considering development of TEMPs, whereas for others who faced these challenges they failed to fully recoup the cost of their investment. In just one example, Smith & Nephew faced difficulties in scaling capabilities to manufacture Dermagraft®, a tissue-engineered skin substitute approved for treating diabetic foot ulcers, at a reasonable cost. Despite achieving FDA approval and showing clinical benefits, Dermagraft has repeatedly failed to turn a profit in part because of its complicated manufacturing requirements.
Compared to cell and gene therapies, TEMPs generally require additional raw materials in the form of cells, scaffolds, and signaling molecules during production. Manufacturers may initially believe that they can easily combine these ingredients based on a pre-determined recipe, but it is often not this straightforward. The quantity and type of raw materials used in production has a significant impact on the end-product. Manufacturers must carefully assess and balance each of the required ingredients and determine how they may impact the validity of an end-product, raising several questions: Which signaling molecules induce appropriate cellular differentiation? Does the scaffold provide a suitable environment for cellular growth and vascularization? Does the end-product mimic the appropriate structural and mechanical properties? The answers to many of these questions remain largely uncertain.
There are also many uncertainties among manufacturers regarding the ability to ensure quality control, consistency, and reproducibility of TEMPs. With limited options for in-line non-destructive testing of critical quality attributes (CQAs), it is often not feasible to accurately assess TEMPs before the production process is complete. TEMPs are also generally manufactured by manually combining cells from the body with scaffold biomaterials, presenting considerable challenges when preparing to scale-up production. This manual production approach is often successfully used when TEMP manufacturers are establishing proof of concept. But as they progress to the commercial level they will need to invest in infrastructures and advanced technologies to be able to rapidly scale-up the production process and ensure end-product quality and reproducibility.
Unclear Regulatory Pathways and Protocols
Another factor limiting the advancement of TEMPs is the lack of clear regulatory pathways for these products compared to those for cell and gene therapies. They also do not appropriately address the added complexity of TEMPs. It is essential that TEMP manufacturers collaborate with the FDA and other regulatory bodies as early as possible in the clinical development process to align on the regulatory requirements for approval. Both parties will need to align on the level of clinical data needed, which may be limited. They will also need to determine the appropriate assessment of manufacturing processes for TEMPs, understanding that there may be few options for in-line, non-destructive testing of CQAs and thus challenges in demonstrating production quality and consistency. If manufacturers could readily demonstrate appropriate levels of quality and consistency, regulatory bodies could more easily differentiate one product from another and make systematic decisions on how best to regulate TEMPs. Many regulatory bodies have made considerable progress in streamlining regulation of TEMPs in recent years, but regulators and manufacturers will need to continue to engage frequently with each other throughout the clinical development process to align on the most effective solutions moving forward.

Suboptimal Reimbursement Strategies
Similar to the lack of clear regulatory pathways and protocols for TEMPs, there are also no universally accepted standards for their reimbursement, especially for TEMPs that offer potentially curative benefit. Curative therapies offer the prospect of long-term benefits with a single or limited number of therapeutic applications, which are valued based on their potentially significant impact on disease severity, quality of life for patients, healthcare system costs, and societal value in terms of increased productivity, reduced hospital visits, and long-term effect on caregivers. Despite any demonstration of long-term value, curative therapies have been scrutinized extensively due to their often-high upfront costs, raising questions about how they should be funded.
Healthcare systems around the world, including nationalized single-payer healthcare systems as seen in Europe and segmented multi-payer healthcare systems such as in the U.S., are currently working to evolve their policies and reimbursement strategies to address the many unique and complex factors associated with curative cell and gene therapies. Industry stakeholders are now collaborating and considering alternative reimbursement models (e.g., cost-sharing, value-based contracts) and/or financing models (e.g., reinsurance, installment payments, consumer health loans) that may better support these therapies. New and emerging reimbursement models being developed for curative cell and gene therapies could potentially be applied to TEMPs in the years ahead to help these often costly and more complex products be effectively valued and reimbursed by global healthcare systems.
The challenges presented by complex manufacturing processes and uncertainties regarding regulatory pathways and reimbursement protocols have led to decreased investor and clinical confidence in TEMPs. Most investors in the life sciences industry understand that there may be uncertainties and risks involved with investigational therapies, but the added complexity of TEMPs and the need for a considerable amount of upfront capital has resulted in a lack of investment in this sector. The associated risks seem to outweigh the promise of innovative TEMPs, producing a bottleneck of novel TEMPs in clinical development stages and reducing the likelihood of commercialization.
But as development and investment in cell and gene therapies continues to advance, learnings from manufacturers and other stakeholders related to these therapies may potentially be applied to the tissue engineering sector to help accelerate development of TEMPs. Stakeholder groups including payers, regulators, and manufacturers must also consider collaborating frequently and early in the clinical development process of TEMPs to align on any regulatory or manufacturing protocols required to achieve regulatory approval and reimbursement. Manufacturers may also need to seek specialized expertise on optimal production strategies that may help to ensure a reliable, reproducible, and consistent manufacturing process, and one that is rapidly scalable at the commercial level.
REFERENCES
- ASTM F2211-13, Standard Classification for Tissue Engineered Medical Products (TEMPs), ASTM International, West Conshohocken, PA, 2013, www.astm.org.
- Dodson BP, Levine AD. Challenges in the translation and commercialization of cell therapies. BMC Biotechnol. 2015;15:70. Published 2015 Aug 7. doi:10.1186/s12896-015-0190-4.
- Gardner, John, and Andrew Webster. 2016. "The social management of biomedical novelty: Facilitating translation in regenerative medicine." Social Science & Medicine 156: 90-97.
- Kim, Y. S., Smoak, M. M., Melchiorri, A. J., & Mikos, A. G. (2018). An Overview of the Tissue Engineering Market in the USA from 2011 to 2018. Tissue Engineering Part A. doi:10.1089/ten.tea.2018.0138.
- Mount, Natalie, Stephen Ward, Panos Kefalas, and Johan Hyllner. 2015. "Cell-based therapy technology classifications and translational challenges." Biological Sciences (The Royal Society Publishing) 307 (1680).
- O’Donnell, B. T., Ives, C. J., Mohiuddin, O. A., & Bunnell, B. A. (2019). “Beyond the Present Constraints That Prevent a Wide Spread of Tissue Engineering and Regenerative Medicine Approaches.” Frontiers in Bioengineering and Biotechnology, 7. doi:10.3389/fbioe.2019.00095.
- Pangarkar, Nitin, et al. “Advanced Tissue Sciences Inc.: Learning from the Past, a Case Study for Regenerative Medicine.” Regenerative Medicine, vol. 5, no. 5, 2010, pp. 823–835., doi:10.2217/rme.10.66.
- Yadav, Vikramaditya, Roza Ghaemi, and Lim Siang. 2019. "Improving the Rate of Translation of Tissue Engineering Products." Advanced Healthcare Materials 8 (19).
About The Authors
The views expressed herein are the authors’ and not those of Charles River Associates (CRA) or any of the organizations with which the authors are affiliated.
Lev Gerlovin is a vice president in the Life Sciences Practice at CRA with more than 12 years’ experience in life sciences strategy consulting, focused on commercial and market access strategies.
Andrew Thomson is a consulting associate within the Life Science Practice at CRA with several years of experience in commercial strategy consulting with pharmaceutical and biotech clients alike.
Jack Vailas is an associate within the Life Sciences Practice at CRA with several years of experience with biotech innovation and commercial strategy.