
Toward A More Robust Regenerative Medicine Regulatory Pathway
By Randy Hubbell, president and CEO, Carmell Therapeutics

In recent months, the U.S. Food and Drug Administration has increased its compliance focus on the regenerative medicine industry, vowing to crack down on illicit stem cell clinics and others advertising the untested use of human cell and tissue-based products (HCT/P), many of which are used in orthopedics.
In December 2018, in a press release regarding its warning letter to a California company marketing unapproved stem cell products, FDA Commissioner Scott Gottleib stated, “The FDA is committed to advancing the field of cell-based regenerative medicine. We’re implementing new policies to make it more efficient to safely develop these promising new technologies. At the same time, we’re also focusing more resources on enforcement when we see companies skirt safety measures and put patients at risk.”
In that same release, the FDA expressed disappointment that very few manufacturers and distributors have taken advantage of its 36-month period of “enforcement discretion,” which began in November 2017 as part of its new policy regarding HCT/Ps that don’t meet all four criteria under 21 CFR 1271.10(a). Stating, “Even though a few sponsors have come to us, we are discouraged by the overall lack of manufacturers wanting to interact with the agency in this enforcement discretion period… [T]here’s a clear line between appropriate development of these products and practices that sidestep important regulatory controls needed to protect patients.”
The FDA further sent To Whom it May Concern letters to some of these regenerative medicine companies stating that, “you appear to be offering “stem cell” products to treat a variety of diseases or conditions” and reiterating the agency’s November 2017 regenerative medicine policy framework.
Medical technology in the sector is advancing rapidly and, in many cases, the industry’s growth is challenging the FDA’s regulatory framework. And while some companies are making false or unsupported claims about the efficacy of their products, there exists a multitude of new products developed by companies that are operating the “right way.”
These companies, in addition to fully meeting the FDA’s regenerative medicine guidelines, are also being responsive to the increasing sophistication of hospitals, group purchasing organizations, and integrated delivery networks, which are heightening their expectations for clinical data to support new products for patient care.
The 351 Vs. 361 Regulatory Framework
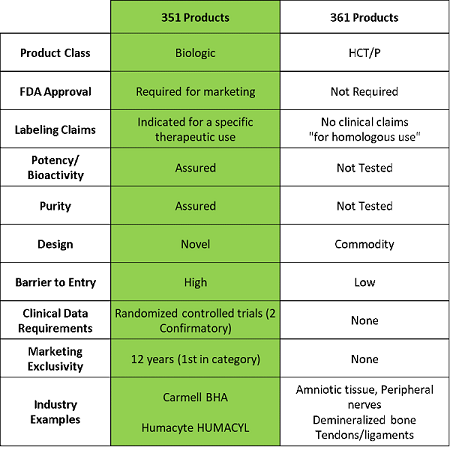
The FDA generally regulates regenerative medicine technologies through Sections 351 and 361 of the Public Health Service Act (PHSA), which is overseen by the Center for Biologics Evaluation and Research (CBER) and gives them the authority to establish requirements for marketing traditional biologics and human cells, tissues, and cellular and tissue-based products. These two regulatory pathways differ markedly in terms of the time, effort, and expense required to bring a product to market in the U.S. Basically, 351 products are regulated as drugs and/or biologics, while 361 products, comparatively, are largely unregulated.
More specifically, blood-derived cell or tissue-based products that fall under the category of 351 products are determined as such because: 1) they are more than minimally manipulated, or 2) they are used for non-homologous work. Manufacturers of these products need to submit a Biologics License Application (BLA) to gain approval in the U.S., which often includes two randomized Phase 3 clinical trials that demonstrate effectiveness on a superiority endpoint and reproducibility at the time of approval.
On the other hand, products that meet the criteria of Section 361 are not subject to any FDA pre-approval process and, as a result, are not required to have any clinical data prior to marketing. In fact, they are subject to few to no FDA requirements with regard to manufacturing and clinical programs. Companies can essentially “self-designate” as meeting the specified criteria and simply need to notify the FDA that they are going to begin marketing and selling to patients. The criteria are as follows:
- The product is minimally manipulated,
- The product is intended for homologous use only (in other words – the tissue has the same function for the recipient as in the donor),
- The manufacture does not involve the combination of the cells or tissues with another article, except for water, crystalloids, or a sterilizing, preserving, or storage agent, and
- The product does not have a systemic effect and is not dependent upon the metabolic activity of living cells for its primary function and is either for autologous use, reproductive use, or allogenic use in a first or second-degree blood relative.
Although the 361 regulatory framework is appropriate for traditional allograft products that are not intended for use as cell-based therapies, many other human tissue products entering the market cannot be considered lawful 361 HCT/Ps because they do not meet the aforementioned four-part test for marketing under this provision.
While stem cells delivered in clinics have been a major driver of the debate between these two regulatory pathways, there are also a host of companies, both public and private, that are manufacturing and selling similar products every day into hospitals, focused on markets such as orthopedics, surgery, and wound-healing. Many of these products are regulated under Section 361, and the companies are making substantial claims about the capabilities of their technology, with no data to support those claims and no regulatory oversight to ensure the existence of manufacturing procedures that guarantee product consistency. Others are offering solutions such as autologous tissue injections but marketing them as stem cells.
The question remains whether these products are following the correct regulatory path. Are they minimally manipulated? Are they truly homologous? Are they being combined with other technologies? These questions unveil a major problem in the marketplace. With stricter interpretation of FDA guidelines on the horizon, many of these products may be at risk of being reclassified to 351s.
And hospitals, physicians, and insurance companies are becoming more skeptical about paying top dollar for 361 products used in surgery, as they lack the clinical data and regulatory rigor necessary to determine that they are providing true benefit to patients. In particular, Value Analysis Committees are looking for more data on products to support cost-risk analysis. They want to see the evidence, compare it to current standards of care, and feel confident in their decision-making.
This ultimately means that the future of the HCT/P industry will likely trend toward the more robust 351 regulatory pathway. Products that undergo this process are entering the marketplace in the way physicians and healthcare executives are looking for: with robust regulatory approval that demonstrates safety and efficacy, supported by successful clinical trials.
About The Author:
 Randy Hubbell is president and CEO of Carmell Therapeutics, a clinical-stage biotechnology company focused on the development and commercialization of innovative regenerative medicine solutions to accelerate bone and soft tissue healing. Hubbell has worked in the life sciences sector for over 25 years, serving in leadership roles at Cardiva Medical, Johnson & Johnson, Enmed, Vasca, Boston Scientific, and IBM. He received his M.B.A. from Loyola University and his B.S. from Tulane University.
Randy Hubbell is president and CEO of Carmell Therapeutics, a clinical-stage biotechnology company focused on the development and commercialization of innovative regenerative medicine solutions to accelerate bone and soft tissue healing. Hubbell has worked in the life sciences sector for over 25 years, serving in leadership roles at Cardiva Medical, Johnson & Johnson, Enmed, Vasca, Boston Scientific, and IBM. He received his M.B.A. from Loyola University and his B.S. from Tulane University.