
The Future Of Gene Therapy Depends On New Delivery Vehicles
By Jonathan Thon, Founder and CEO, STRM.BIO

Gene therapy has the potential to cure our most serious diseases and extend human lifespans by modifying the basic building blocks of life to restore or enhance cell function. Despite its significant promise, the field has yet to meet its full potential. The few therapies that have made it to market so far have been for rare, single-gene diseases, and the treatments are challenging; gene therapies are predominantly delivered ex vivo, an approach in which a patient’s cells are edited in culture and then re-transfused. Like most transplant procedures, the patient must undergo immune system ablation before receiving an ex vivo gene therapy treatment, which can have severe side-effects or even be fatal1. Administration requires specialized medical training and hospitalization in dedicated facilities, contributing to costs that are too high to be supported by payers as a routine option2-4. This approach is not sustainable, and thus ripe for disruption.
The field has already overcome several bottlenecks. The first was to identify the genetic variants responsible for a given disorder, which used to be a very slow and laborious process. In the era of big data and genomics, target identification is now relatively fast and straightforward. While the first generation of gene therapies in the 1990s essentially involved delivering a functional gene sequence into cells, new precision editing technologies such as CRISPR–Cas9, base editing, and prime editing mean that making the desired modifications to the DNA is no longer a technical bottleneck. Modern multiplexed therapies that can edit several genes in parallel mean that the applications of gene therapy can expand beyond relatively rare single-gene inherited disorders for the first time, opening the possibility of treating common complex diseases such as cancer or diabetes.
And yet the list of approved gene therapies remains short, while the risks and costs of the current therapies remain high.
The final bottleneck is the safe and efficient delivery of gene therapeutic constructs in vivo. Consider an analogy from the computing world. It’s like trying to get high-performance cloud computing into home computers via dial-up modems, while waiting for someone to invent broadband. Solving the gene therapy delivery problem by enabling direct injection and precision targeting will transform the field by helping bring new treatments and cures for diverse diseases into every hospital5.
The ideal gene therapy delivery vehicle should meet the following criteria:
- Safe. Patient safety is paramount, and gene therapy vehicles that cause severe side effects are not a sustainable option. As part of this safety criterion—as well as regulators’ chemistry, manufacturing, and controls (CMC) requirements—delivery vehicles must have consistent physical and chemical characteristics and quality.
- Repeatable, in vivo. It is critical that the next generation of gene therapies be implemented as simple injections in standard clinical settings, without the need for risky immune system ablation and the associated long hospital stays. Since some gene therapies will require more than one dose, the ideal delivery vehicle for in vivo applications will need to have immune privilege. Without it, the immune system will recognize the first dose of the therapy as an external threat in the same way that it would recognize an infectious disease, triggering a response that attacks and destroys subsequent doses, limiting therapies to one-and-done treatments.
- High capacity. The increasing precision and multiplexing capability of modern gene therapy technology often mean a larger cargo, including not just DNA or RNA molecules but also proteins that catalyze their use within the cell. The ideal delivery vehicle will be capable of encapsulating and delivering diverse types of gene therapeutic payloads.
- Targetable. Treatments for different diseases will require different cell type specificity (cell tropism). Long-acting treatments and permanent cures will generally need to target stem and progenitor cells.
A solution that meets all these criteria will democratize gene therapy by providing a simpler and safer way to deliver treatments and cures as standard of care for diverse diseases.
The Trouble with Viruses
The gene therapy delivery market has been dominated by viruses since the dawn of the field, thirty years ago. The initial attraction of viruses is clear: they can deliver DNA/RNA cargos into human cells; they come in different sizes suitable for a range of cargo types; and they have innate (if broad) serological tropism. But they also have meaningful drawbacks.
The primary disadvantages of viral vectors relate to repeatability and safety. The human immune system has evolved to mount a strong response to viral vectors, posing a major challenge for gene therapies that require repeat dosing in vivo6. The immune response can also cause side effects, some of which can be serious, even fatal. Other safety concerns around viral vectors include insertional mutagenesis that can activate oncogenes and have other unintended effects7-9, and unanticipated and unpredictable molecular changes during production that can create an inconsistent product10,11.
Viral vectors also do not have sufficiently targeted cell tropism and biodistribution for many in vivo applications. For example, vectors based on retroviruses and lentiviruses can only infect dividing cells, while adenovirus vectors can be taken up by any cell type, increasing both the risk of side effects and the effective dose.
Although many research teams are still working to develop improved versions of viral delivery vehicles for gene therapies, the future of the field likely lies elsewhere. There are several candidates to replace viruses as the delivery vehicle of choice for gene therapies, each with advantages and disadvantages with respect to the criteria laid out above.
Synthetic Nanoparticles
Synthetic nanoparticles based on lipids, proteolipids, and other polymers were developed to engineer a safer and more targetable version of a natural viral vector. Lipid nanoparticles (LNPs) are the most familiar class, having gained prominence during the COVID-19 pandemic as the vehicle used to deliver the mRNA vaccines produced by Pfizer–BioNTech and Moderna, but there are several versions of this technology with different characteristics.
Repeatability and Safety: Synthetic nanoparticle technology went through several iterations prior to initial clinical approval, and trade-offs have been made between delivery efficiency, short-term safety, and immune system response12. The current generation of synthetic particles has a better safety profile than viral vectors, although a small percentage of the population may mount an immune, inflammatory, or (very rarely) even an allergic response to the ingredients of some formulations13. Nevertheless, synthetic particles exhibit better immune privilege than viral vectors for most individuals, permitting a limited number of repeat doses14. On the CMC front, a consistent synthetic nanoparticle product can be delivered at scale. Public acceptance of LNPs is generally high due to their recent successful use in COVID-19 vaccines.
Capacity: LNPs have a medium-sized carrying capacity, broadly equivalent to that of lentiviral vectors, while proteolipid vehicles (PLVs) can encapsulate larger cargos, about midway between that of lentiviruses and adenoviruses15. They can thus encapsulate most—but not all—current cargo types, and new generations of larger particles may be needed as cargo size continues to increase.
Targetability: While synthetic particles have no innate cell tropism, they accumulate largely in the liver and as such have strong potential to deliver gene therapies for liver diseases. Other cell tropisms and biodistribution profiles can be engineered to some extent through chemical or electrostatic modification or the incorporation of cell surface proteins. However, some of these modifications can increase toxicity and reduce immune privilege while introducing CMC-related challenges. Since natural cell tropism is driven by the recognition of complex combinations of cell surface proteins—less like a single key fitting a lock, and more like a secret handshake—this limited engineered tropism is typically suitable only for a few specific cell types and can be unpredictable in vivo.
Biologically-Derived Particles
Biologically-derived particles can be subdivided into two distinct subtypes that may be adoptable as gene delivery vehicles: exosomes, which derive from inside cells and constitute the basis of the first generation of non-viral gene delivery companies that emerged in the first part of the decade, and microvesicles, which bud from the cell surface membrane and are easier to characterize, but are ironically newer entrants into this space. As part of the body’s intercellular communication network, these extracellular vesicles (EVs) have an innate ability to encapsulate human RNAs, DNAs, and proteins and to efficiently deliver this cargo into other cells—several orders of magnitude more efficiently than LNPs and other synthetic particles16.
Much of the early work on EVs as therapies focused on exosomes derived from mesenchymal stem cells, as these were easily accessible and can recapitulate some of the regenerative and other beneficial properties of the stem cells themselves17. In parallel, researchers developing systems for the large-scale manufacture of blood cells and blood products from cultured cells also began to understand the role of extracellular vesicles in transferring nucleic acids and proteins between hematopoietic cells. Thanks to this foundational work, exosomes and microvesicles recently entered development as delivery vehicles for gene therapy, especially for applications targeting hematopoietic stem and progenitor cells18-21.
A comparison of exosomes and microvesicles is shown in Figure 1.
Figure 1. The characteristics of microvesicles provide distinct advantages for in vivo delivery of gene therapies when compared to exosomes.
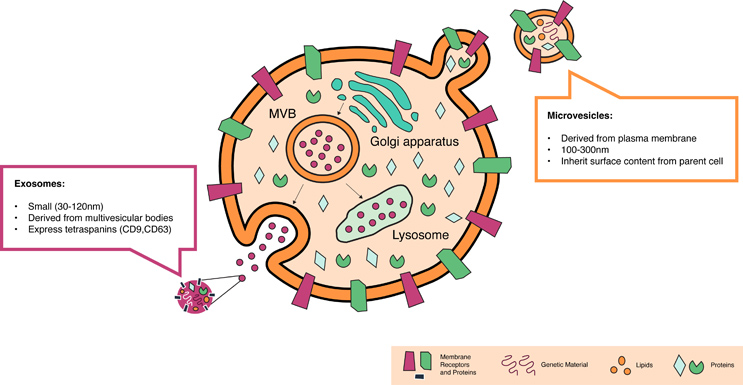
Repeatability and Safety: EVs are natural products generated by almost every cell in the body, and thus have significant safety and immune privilege advantages over viral and synthetic particles, allowing more repeat doses in vivo with fewer side effects. As EVs are naturally secreted into the blood and other bodily fluids from multiple types of cells22-24, they have been present in every blood transfusion and organ transplantation ever performed, beginning long before their formal discovery and characterization; they’re also present in other biological products. In addition, the body is thought to naturally use EVs to repackage and transport cargo delivered into cultured cells by LNPs and other synthetic particles, for example delivering mRNA from muscle cells at a vaccine injection site to immune system cells elsewhere in the body25. This long history demonstrates their safety.
Both forms of EVs can be readily manufactured at scale from liquid cell culture. The small size of exosomes does create challenges around product characterization and quality control, since conventional analysis tools cannot accurately characterize particles smaller than ~100 nm in size. Exosomes will therefore continue to face significant CMC-related barriers to regulatory approval until improved analytical tools become available. In contrast, microvesicles do not share this limitation and can be manufactured with more consistent properties and at larger scales.
Capacity: Exosomes are very small, with a size roughly equivalent to that of adeno-associated virus vectors, the smallest member of the viral vector category. This limits the types of cargo they can carry. Microvesicles are larger, with a carrying capacity similar to and likely greater than adenoviruses. As such they can be used to deliver a diverse range of cargos, including the larger constructs required for multiplexed gene editing applications26-29.
Targetability: Exosomes have limited known surface biomarkers and therefore suffer from the same cell tropism issues as LNPs and other synthetic vesicles, although again as with LNPs a certain degree of tropism can be engineered. In contrast, when microvesicles bud from the cell surface membrane they inherit the complex combination of surface proteins expressed by their parent cell. Microvesicles derived from different cell types therefore have different cell tropisms, making them a highly tunable option for diverse gene therapy applications. Select microvesicles can naturally home to hematopoietic stem cells in the bone marrow and other locations, offering the prospect of long-lasting in vivo treatments and even cures for a range of inherited hematopoietic disorders. Some microvesicles even have the innate ability to cross the blood-brain barrier, potentially allowing access to cell types that are extremely challenging to target using other means30. Cultured source cells can be genetically engineered to knock out or modify any cell surface proteins as desired, offering further, more specific and tunable cell tropism than is possible with other delivery vehicle types.
Hybrid Vehicles
Sharing the advantages and disadvantages of both, hybrid delivery vehicles are produced by fusing synthetic nanoparticles with EVs. Because they are larger, hybrid particles have a larger carrying capacity than LNPs/exosomes and may offer better targetability, at the expense of synthetic components that are likely to cause similar safety and immunogenicity challenges to those observed with LNPs.
The Future is Bright
This is an exciting time for our field! Solving gene delivery will make lifesaving gene therapies a reality for those in need. As novel delivery modalities mature, the gene therapy ecosystem is on the cusp of a transformative breakthrough and should be watched closely for the surge of opportunities that is to come.
About the Author:
A serial biotech entrepreneur, Jonathan Thon is the founder and CEO of STRM.BIO, a pre-clinical, VC-backed biotechnology company that is leveraging extracellular vesicles (EVs) to deliver gene therapies. Prior to launching STRM.BIO he founded and served as CEO / CSO of PlateletBio, where he helped develop next-generation allogeneic cell therapies for the treatment of human diseases.
References:
- Al-Saif, A. M. (2019). Gene therapy of hematological disorders: current challenges. Gene Ther, 26(7-8), 296-307. https://doi.org/10.1038/s41434-019-0093-4
- Joszt, L. (2021, May 19). Gene Therapies Present Reimbursement Challenges That Have Yet to Be Answered. The American Journal of Managed Care. https://www.ajmc.com/view/gene-therapies-present-reimbursement-challenges-that-have-yet-to-be-answered
- Doxzen, K. (2021, August 13). New gene therapies may soon treat dozens of rare diseases, but million-dollar price tags will put them out of reach for many. The Conversation. https://theconversation.com/new-gene-therapies-may-soon-treat-dozens-of-rare-diseases-but-million-dollar-price-tags-will-put-them-out-of-reach-for-many-164990
- Durante, R. (2020, January 6). Solving the Gene Therapy Cost-Effectiveness Conundrum. Maru Group. https://www.marugroup.net/insights/blog/gene-therapy-cost-effectiveness
- LaMotte, S. (2022, May 31). Changing our DNA: 'The age of human therapeutic gene editing is here.’ CNN. https://www.cnn.com/2022/05/31/health/reversing-genetic-fate-scn-wellness/index.html
- Verdera, H. C., Kuranda, K. & Mingozzi, F. (2020). AAV Vector Immunogenicity in Humans: A Long Journey to Successful Gene Transfer. Mol Ther 28(3), 723-746. https://doi:10.1016/j.ymthe.2019.12.010
- Rothe, M., Modlich, U. & Schambach, A. (2013). Biosafety challenges for use of lentiviral vectors in gene therapy. Curr Gene Ther 13(6), 453-468. https://doi:10.2174/15665232113136660006
- Check, E. (2002). Regulators split on gene therapy as patient shows signs of cancer. Nature 419, 545-546. https://doi:10.1038/419545a
- Goswami, R. et al. (2019). Gene Therapy Leaves a Vicious Cycle. Front Oncol 9(1), 297. https://doi:10.3389/fonc.2019.00297
- McCarron, A., Donnelley, M., McIntyre, C. & Parsons, D. (2016). Challenges of up-scaling lentivirus production and processing. J Biotech 240, 23-30. https://doi:10.1016/j.jbiotec.2016.10.016
- Giles, A. R. et al. (2018). Deamidation of Amino Acids on the Surface of Adeno-Associated Virus Capsids Leads to Charge Heterogeneity and Altered Vector Function. Mol Ther 26(12), 2848-2862. https://doi:10.1016/j.ymthe.2018.09.013
- Cross, R. (2021, March 6). Without these lipid shells, there would be no mRNA vaccines for COVID-19. Chemical & Engineering News. https://cen.acs.org/pharmaceuticals/drug-delivery/Without-lipid-shells-mRNA-vaccines/99/i8
- Mohamed, M. et al. (2019). PEGylated liposomes: immunological responses. Sci Technol Adv Mater 20(1), 710-724. https://doi.org/10.1080/14686996.2019.1627174
- Kenjo, E. et al. (2021). Low immunogenicity of LNP allows repeated administrations of CRISPR-Cas9 mRNA into skeletal muscle in mice. Nat Commun 12, 7101. https://doi:10.1038/s41467-021-26714-w
- Lundstrom, K. (2018). Viral Vectors in Gene Therapy. Diseases 6(2), 42. https://doi.org/10.3390/diseases6020042
- Murphy, D. E. et al. (2021). Natural or Synthetic RNA Delivery: A Stoichiometric Comparison of Extracellular Vesicles and Synthetic Nanoparticles. Nano Lett 21 (4), 1888-1895. https://doi:10.1021/acs.nanolett.1c00094
- Zhao, A. G., Shah, K., Cromer, B. & Sumer, H. (202). Mesenchymal Stem Cell-Derived Extracellular Vesicles and Their Therapeutic Potential. Stem Cells Int 2020, 8825771. https://doi:10.1155/2020/8825771
- Bunggulawa, E. J. et al. (2018). Recent advancements in the use of exosomes as drug delivery systems. J Nanobiotechnology 16, 81. https://doi:10.1186/s12951-018-0403-9
- Kao, C. Y. & Papoutsakis, E. T. Extracellular vesicles: exosomes, microparticles, their parts, and their targets to enable their biomanufacturing and clinical applications. Curr Opin Biotechnol 60, 89-98. https://doi:10.1016/j.copbio.2019.01.005
- Wiklander, O. P. B., Brennan, M. A., Lotvall, J., Breakefield, X. O. & El Andaloussi, S. (2019). Advances in therapeutic applications of extracellular vesicles. Sci Transl Med 11(492). https://doi:10.1126/scitranslmed.aav8521
- Kao, C. Y. & Papoutsakis, E. T. (2018). Engineering human megakaryocytic microparticles for targeted delivery of nucleic acids to hematopoietic stem and progenitor cells. Sci Adv 4(11), eaau6762. https://doi:10.1126/sciadv.aau6762
- Butler, J. T., Abdelhamed, S. & Kurre, P. (2018). Extracellular vesicles in the hematopoietic microenvironment. Haematologica 103(7), 382-394. https://doi:10.3324/haematol.2017.183335
- Camussi, G., Deregibus, M. C., Bruno, S., Cantaluppi, V. & Biancone, L. (2010). Exosomes/microvesicles as a mechanism of cell-to-cell communication. Kidney Int 78(9), 838-848. https://doi:10.1038/ki.2010.27
- Mause, S. F. & Weber, C. (2010). Microparticles: protagonists of a novel communication network for intercellular information exchange. Circ Res 107(9), 1047-1057. https://doi:10.1161/CIRCRESAHA.110.226456
- Maugeri, M. et al. (2019). Linkage between endosomal escape of LNP-mRNA and loading into EVs for transport to other cells. Nat Commun 10, 4333. https://doi:10.1038/s41467-019-12275-6
- Kao, C. Y. & Papoutsakis, E. T. (2018). Engineering human megakaryocytic microparticles for targeted delivery of nucleic acids to hematopoietic stem and progenitor cells. Sci Adv 4(11), eaau6762. https://doi:10.1126/sciadv.aau6762
- Lamichhane, T. N., Raiker, R. S. & Jay, S. M. (2015). Exogenous DNA Loading into Extracellular Vesicles via Electroporation is Size-Dependent and Enables Limited Gene Delivery. Mol Pharm 12(10), 3650-3657. https://doi:10.1021/acs.molpharmaceut.5b00364
- O'Loughlin, A. J. et al. (2017).Functional Delivery of Lipid-Conjugated siRNA by Extracellular Vesicles. Mol Ther 25(7), 1580-1587. https://doi:10.1016/j.ymthe.2017.03.021
- Pomatto, M. A. C. et al. (2019). Improved Loading of Plasma-Derived Extracellular Vesicles to Encapsulate Antitumor miRNAs. Mol Ther Methods Clin Dev 13, 133-144. https://doi:10.1016/j.omtm.2019.01.001
- Banks, W. A., Sharma, P., Bullock, K. M., Hansen, K. M., Ludwig, N., & Whiteside, T. L. (2020). Transport of Extracellular Vesicles across the Blood-Brain Barrier: Brain Pharmacokinetics and Effects of Inflammation. International journal of molecular sciences, 21(12), 4407. https://doi.org/10.3390/ijms21124407