
Solving Bioassay Challenges for Cell & Gene Therapies
By Minkyung Kim, Pharm.D., MBA, M.S., USP; Nikhil Rautela, M.Sc., M.Phil., USP; Shubhrata Khedkar, M.S., USP

To foster the development and licensing of quality advanced therapies, the Asia-Pacific Economic Cooperation (APEC) has established Training Centers of Excellence (CoE) for regulatory science within several priority work areas. One of these areas is advanced therapies, and the United States Pharmacopeia (USP) was designated a CoE for Advanced Therapies by the APEC Regulatory Harmonization Steering Committee following a successful pilot CoE training program on raw materials for advanced therapies in 2021. This January, the USP CoE held a bioassay training workshop that included regulatory considerations, analytical validation, and case studies related to bioassays for cell and gene therapy (CGT) products. This training aimed to address the development and validation of bioassays, which play a key role in assessing potency. Potency is a critical quality attribute (CQA) that should tie to the therapeutic’s mechanisms of action (MOA) and should link clinical efficacy to dosage.1
Potency Assays
21 CFR 600.3(s) defines potency as “the specific ability or capacity of the product, as indicated by appropriate laboratory tests or by adequately controlled clinical data obtained through the administration of the product in the manner intended, to effect a given result.”2 Although there are various international guidelines to facilitate compliance with regulatory expectations for potency assays (examples are shown in Figure 1), the complexity of the MOA of CGT products and absence of suitable standards remain unique challenges to developing potency assays for CGT products.
There are different types of bioassays to quantitatively measure biological properties, such as animal-based, cell-culture-based, and biochemical assays.3 Even though animal-based assays are often most relevant to clinical response, cell-based bioassays are regarded as the most suitable for potency determination due to their lower variability compared to animal-based assays as well as ethical concerns. Therefore, manufacturers are often advised to limit the use of animals whenever possible.4
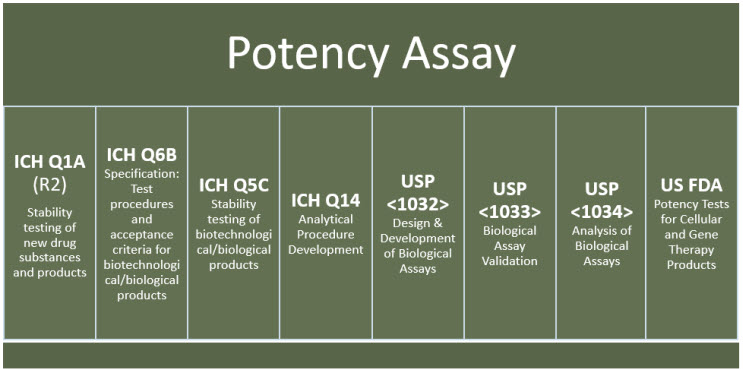
Figure 1. Examples of international resources and best practices for potency assay
Regulatory Considerations
Ramjay Vatsan, from FDA’s Center for Biologics Evaluation and Research (CBER), shared that a good potency assay reflects the mode of action and is reproducible, accurate, robust, stability-indicating, and practical. Vatsan emphasized the importance of exploring orthogonal methods. Assays should include system suitability controls and be validated for accuracy, detection limit, precision (i.e., repeatability and intermediate precision), specificity, linearity and range, and robustness. He also discussed scientific considerations during the development of potency assays for CAR-T cells, such as, for example, identifying the product attributes that reflect product performance and selecting the most appropriate cell-based potency assay (e.g., cytokine production, proliferation, lytic activity). He shared a potency assay case study for a cancer vaccine that used an enzyme-linked immunosorbent assay (ELISA) kit to measure serum cytokine concentrations from mice injected with various product dilutions. He explained that even though the manufacturer was directly measuring product activity that was relevant to the MOA, the ELISA kit was measuring a subunit common to multiple cytokines, not just the one of interest, so it may not be appropriate. He stressed the importance of knowing what the assay is measuring and understanding the underlying biology.
Yoji Sato from Japan’s National Institute of Health Sciences expanded on regulatory guidelines by referring again to the 21 CFR 610.10 requirements regarding potency and emphasizing that for advanced therapies, multiple complementary assays can be particularly valuable.5 This approach, often called a matrix approach, can include quantitative and/or qualitative assays, but qualitative assays should complement one or more quantitative assays for lot release, stability, or comparability studies.4
Implementing A Life Cycle Approach To Expedite Development And Approval
Various speakers mentioned the CGT product life cycle and a matrix approach, especially since many therapeutic candidates qualify for expedited regulatory programs. The conventional approach of postponing assay development and characterization until clinical studies can delay licensure; therefore, manufacturers should stipulate the performance requirements and explore orthogonal methods and matrix assays as early as possible to expedite product development.
It is important to design and establish an assay profile and feasibility criteria early in the development process — including considerations for a gene of interest, MOA, and biological function — and to assess if multiple assays for expression and functionality are required. Design should consider the limitations in the understanding of final manufacturing processes, differences between drug substance and drug product, and limited sample size.6
Developing a relative cell-based potency assay is a common approach to assigning potency but is dependent on an appropriate biologically active and relevant reference standard. Once the ideal reference standard profile is established, it helps guide assay development by ensuring that appropriate controls, replicates, and statistics demonstrate all the required assay parameters. Vatsan recommended early assay qualification studies for all assays and, where feasible, performing early assay validation.
Adeno-associated Viral Vectors: Case Study
Linda Engle of Biogen presented a case study of developing a cell-based potency bioassay for gene therapy products based on adeno-associated viral (AAV) vectors. She elaborated that potency assays for AAVs can be based on several biological processes, such as vector infectivity, transgene mRNA expression, transgene protein expression, and transgene protein function. Assays should be fit for purpose. Engle discussed the following key steps and considerations involved in developing an immunoassay to measure transgene protein expression:
- Selecting cell lines — tissue-specific promotor requirements, the permissiveness of viral infection, cellular components required for transgene expression, ease of growth, cell line stability in culture, optimization via cell line engineering
- Optimizing infection and cell lysis conditions
- Developing a protein detection system — signal-to-noise and lysis buffer interference
- Analyzing edge effects — plate layout and uniformity across the plate
- Analyzing data using an appropriate statistical model.
Common criteria used to measure similarity in cell-based bioassays include:
- Ratio of slopes from the unconstrained model
- Ratio of lower or upper asymptotes from the unconstrained model
- The difference between the upper and lower asymptotes from the constrained model.
Table 1. Widely used models for in vitro bioassays

Bioassay Considerations
To introduce a potency assay into a GMP environment, it must be statistically robust and provide objective measurements. It is widely recommended to perform a design of experiments (DOE) when qualifying and validating methods, during which phase-appropriate assay criteria are established based on sound statistical analysis and more stringent criteria are developed for products in later phases of development.
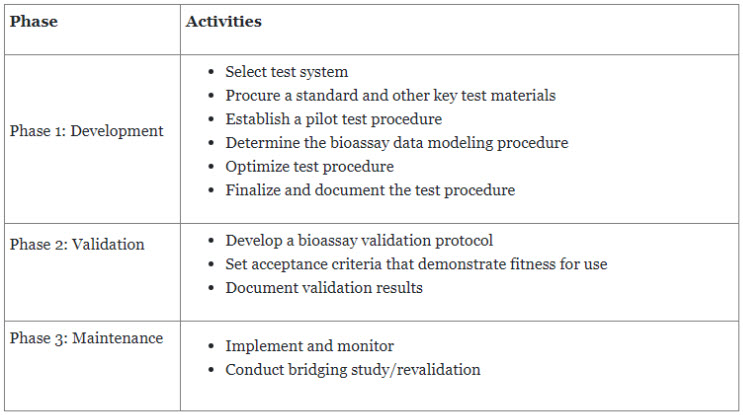
Steven Walfish from USP discussed the life cycle of bioassays around the continuous improvement approach for design and development, procedure performance qualification, and continuous verification, which is also discussed in USP General Chapter <1220> Analytical Procedure Life Cycle7 and forthcoming ICH Q14. Although it is continuous, the bioassay life cycle is considered to consist of three major phases, as described in Table 2.
Table 2. Three major phases of the bioassay lifecycle
Chae-Ok Yun, from GeneMedicine Co. and Hanyang University, presented a case study on the development and validation of a method based on optical density that measures infectious adenovirus particles by tissue culture infectious dose 50% (TCID50). The effect of plate design was discussed. Because the assay requires incubation of 14 days, edge dryness at the outer columns of a 96-well plate was studied, and once it was confirmed that there was an edge effect, the extreme columns were excluded from the final plate map, highlighting the importance of plate design and the impact it can have on data.
Bioassay Validation
USP General Chapter <1033> Bioassay Validation reviews the fundamentals and provides an example of bioassay validation.8 Bioassay validation design should consider all facets of the measurement process. The goal of validation is to confirm the operating characteristics of the procedure to demonstrate that the bioassay is suitable for its intended use.8 Sources of bioassay measurement variability include sample preparation, intra-run factors, and inter-run factors. Table 3 further elaborates on the validation parameters, which are also covered in USP General Chapter <1225> Validation of Compendial Procedures.
Table 3. Method Validation Parameters
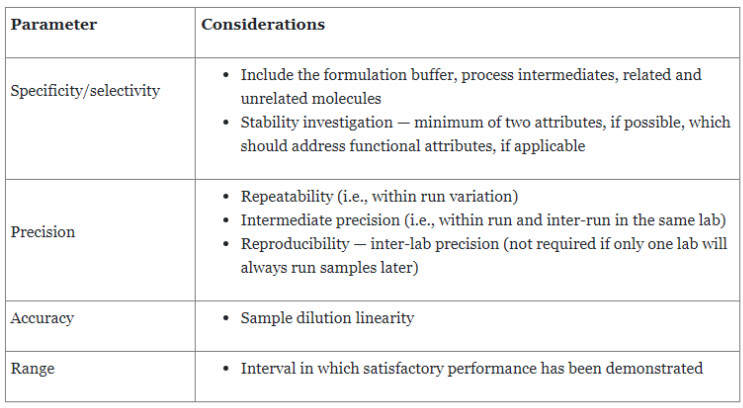
Precision, accuracy, and range can be efficiently addressed through a common experimental setup. Precision is usually expressed as relative standard deviation (RSD), whereas accuracy is expressed as the percentage of recovery. An important step before designing a bioassay is the pre-validation assessment, which should consider the following questions:
- Is the equipment suitable for the expected accuracy?
- Are the reference materials suitably characterized?
- Is the analytical method procedure finalized?
- Has the validation protocol been approved by management and quality assurance?
A bioassay often uses relative potency and statistical models to determine a reportable value. The statistical elements of bioassay development include the type of data, the measure of response at varying concentrations, the assay design and statistical model, pre-analysis treatment of the data, methods of data analysis, suitability testing, and outlier analysis.7 As noted in USP General Chapter <1032> Design and Development of Biological Assays, biological systems have inherent variability from a variety of sources, including animals, cells, instruments, reagents, time, and location; therefore, an absolute measure of cell-based bioassay potency is more variable than a measure of activity relative to a standard.9 Standards for relative potency can be used to support potency measurements, trending, and stability testing.
The APEC-USP CoE bioassay for advanced therapies training is part of USP’s ongoing efforts to foster regulatory convergence to support greater access to innovative therapeutics. Defining a suitable cell-based potency bioassay strategy can help mitigate challenges, accelerate development pathways, and remove inconsistencies that can raise regulatory concerns. In hopes of spreading this work more broadly, USP has made this training available to any interested stakeholder free of charge through this link (you will need USP Access Point login credentials to access). USP continues to work closely with regulators and other stakeholders to provide best practices and tools for priority areas such as advanced therapies.
References
- Pimpaneau V, Gianelli F, Trouvin JH, Poiseau AD. The challenges of potency assay development for cell-based medicinal products in Europe. Regulatory Rapporteur. 2015; 12(5): 5-10.
- https://www.accessdata.fda.gov/scripts/cdrh/cfdocs/cfcfr/CFRSearch.cfm?fr=610.10
- ICH. Q6B: Specifications: Test procedures and acceptance criteria for biotechnological/biological products.
- US FDA. Potency tests for cellular and gene therapy products. 2011. https://www.fda.gov/files/vaccines,%20blood%20&%20biologics/published/Final-Guidance-for-Industry--Potency-Tests-for-Cellular-and-Gene-Therapy-Products.pdf
- https://www.accessdata.fda.gov/scripts/cdrh/cfdocs/cfcfr/cfrsearch.cfm?fr=600.3
- Liloia C, Ziehr B, McVicker D, Wong YY, Carrillo E, Rodriguez A. Analytical strategies for cell and gene therapy development. BioPharm International. 2021;34(6):42-45.
- USP. General Chapter <1220> Analytical procedure life cycle. USP–NF 2022 Issue 1. DOI: https://doi.org/10.31003/USPNF_M10975_02_01
- USP. General Chapter <1033> Bioassay validation. USP–NF 2022 Issue 1. DOI: https://doi.org/10.31003/USPNF_M912_01_01
- USP. General Chapter <1032> Design and development of biological assays. USP–NF 2022 Issue 1. DOI: https://doi.org/10.31003/USPNF_M1354_01_01
About The Authors:
 Minkyung Kim, Pharm.D., MBA, M.Sc., is a scientific affairs manager at USP, APAC. She holds her licenses as a registered pharmacist in the state of Pennsylvania and South Korea. Kim leads global cell and gene landscaping projects to identify opportunities and challenges for product development; distinguish global, regional, and local key opinion leaders; and create a network for further collaboration. Before joining the USP, she worked at Mundipharma and Bayer in medical affairs, where she led advisory board meetings for immunology and oncology products and deployed diagnostic validating platforms for various companion diagnostics. Her work experience also includes research at the National Cancer Center in South Korea. You can reach her on LinkedIn.
Minkyung Kim, Pharm.D., MBA, M.Sc., is a scientific affairs manager at USP, APAC. She holds her licenses as a registered pharmacist in the state of Pennsylvania and South Korea. Kim leads global cell and gene landscaping projects to identify opportunities and challenges for product development; distinguish global, regional, and local key opinion leaders; and create a network for further collaboration. Before joining the USP, she worked at Mundipharma and Bayer in medical affairs, where she led advisory board meetings for immunology and oncology products and deployed diagnostic validating platforms for various companion diagnostics. Her work experience also includes research at the National Cancer Center in South Korea. You can reach her on LinkedIn.
 Nikhil Rautela, M.Sc., M.Phil., is a senior scientific affairs manager for biologics at USP India. His primarily role is to engage with stakeholders to advocate for the quality of USP science supporting biologics as well as peptides and oligonucleotides in the EMEA region. You can reach him on LinkedIn.
Nikhil Rautela, M.Sc., M.Phil., is a senior scientific affairs manager for biologics at USP India. His primarily role is to engage with stakeholders to advocate for the quality of USP science supporting biologics as well as peptides and oligonucleotides in the EMEA region. You can reach him on LinkedIn.
 Shubrata Khedkar is the bioassay lead at USP India’s biologics lab. She is also an approved instructor for the USP education courses, “Bioassay-Design, Development, and Validation” and “Residual DNA Testing”. You can reach her on LinkedIn.
Shubrata Khedkar is the bioassay lead at USP India’s biologics lab. She is also an approved instructor for the USP education courses, “Bioassay-Design, Development, and Validation” and “Residual DNA Testing”. You can reach her on LinkedIn.