
SARS-CoV-2: Massey Univ. Profs Develop Method To Quickly Sequence Genome
By Nikki E. Freed, Olin Silander, and Nick Downey

With more than 12,000 mutations occurring in the SARS-CoV-2 virus since it was discovered,1 it was perhaps simply a matter of time before the world saw variants emerge that increased the transmission rate of COVID-19. Next-generation sequencing (NGS) has been vital for not only the sequencing of the viral genome but for keeping track of changes in it. A research team led by two of us (Nikki Freed and Olin Silander, at Massey University, New Zealand) have developed a new NGS method, called the Midnight Panel, for the ultrafast sequencing of the SARS-CoV-2 genome, which cuts the sequencing time by more than half compared with more standard methods.2
Sequencing Viral Genomes
Not only does the sequencing of viruses allow scientists to monitor for changes in their genome, but such detection also facilitates the mapping of the various mutations and their relationships by building phylogenetic trees. Sequencing also enables the determination of evolution rates, identification of transmission routes, contact tracing, and population surveillance. Viral genomic data also informs the researchers that create diagnostic tests and the research on vaccines and antiviral therapies.
Since the emergence of COVID-19, several NGS methods have been used to sequence the SARS-CoV-2 virus that causes the disease; the most common one is the protocol developed by Joshua Quick and his colleagues in the ARTIC (Advancing Real-Time Infection Control) Network.3-5 The SARS-CoV-2 ARTIC method employs a targeted approach, sequencing only the viral genome and not the entire RNA sample, which includes a huge excess of human RNA (the human transcriptome).
There are several advantages to using the targeted sequencing rather than the whole transcriptome sequencing (WTS) approach. First, targeted sequencing increases the read depth for low-quantity samples of RNA, which increases the likelihood of identifying rare variants. Targeted approaches also tend to be faster and more cost-effective.
The general ARTIC method was initially developed for the genomic surveillance of the Ebola virus during an outbreak in a region where access to advanced NGS technologies was limited. As such, the ARTIC method employed a portable sequencer—revolutionary at the time—to provide real-time, mobile sequencing.6 Freed and Silander adapted the Ebola surveillance method and ARTIC method to decode the SARS-CoV-2 genome. Called the Midnight Panel, the method facilitates the global effort to understand COVID-19 epidemiology.2
NGS With The Midnight Assay
The Midnight Panel, like the ARTIC method, adopts a targeted sequencing approach to increase the speed, quality, and cost-effectiveness of SARS-CoV-2 genome surveillance. The first step in the Midnight Panel is the extraction of RNA from virus-positive samples. RNA is extracted instead of DNA because coronaviruses (such as SARS-CoV-2) have a genome made up of RNA. To stabilize the sample and prepare it for sequencing, the RNA is converted into its complementary DNA (cDNA) sequence.
The next step of the Midnight protocol is the amplification of viral cDNA by the polymerase chain reaction (PCR). Using a pair of short DNA strands, called primers, which complement either end of the selected SARS-CoV-2 sequence, and a thermostable enzyme, DNA polymerase, small areas of the viral genome are “amplified” or copied. Each time a copy is created, it serves as a template for more copies. Thus, after only a few cycles, many copies of the selected region of the SARS-CoV-2 cDNA viral genome are generated. To save time and money, different primer pairs can be used simultaneously in what is referred to as a multiplex PCR, thus enabling many copies of the entire SARS-CoV-2 genome to be amplified or copied.
For sequencing, the primer pairs are designed to produce amplicons that overlap by 50 to 100 base pairs (bp), so that after the amplicons have been sequenced, the overlapping sequences can be lined up to construct the genome sequence. Overlapping or tiled amplicons also ensure the primers themselves do not “hide” mutations. With the Midnight Panel, the assembly of the genome is guided by comparison against a reference SARS-CoV-2 sequence. Since tiled amplicons have overlapping regions, the PCR step is run with two separate sets of primers—one set generating odd-numbered amplicons and the other set generating even-numbered amplicons. These resulting two pools of amplicons are then combined before sequencing (Figure 1).

Figure 1. SARS-CoV-2 Midnight Panel includes 30 primer pairs in pool 1 that amplify the odd regions of SARS-CoV-2 and 28 primer pairs in pool 2 that amplify the even regions. Adapted from Figure 3. Genome coverage plots for patient samples varying in Cq values in [2]. (Licensed under CC BY 4.0)
Finally, each amplicon is sequenced hundreds, if not thousands, of times. The data must then be assembled by comparing each amplicon sequence to a reference genome sequence. Since many copies are generated from the individual segments of the viral genome, copying errors can occur. The more copies of each amplicon that are sequenced, the more likely it is to identify a true viral variant rather than a copying error. The data for each amplicon or copy is called a read, and the number of reads that align to a segment of the genome is sequencing depth or read depth. The genome sequences and reference sequence can then be compared to identify any differences or “variants,” in single letters of the genome (the nucleotide). This process allows us to decode the sequence of entire 30,000 bp of the viral genome.
Benefits Of The Midnight Panel
While much more efficient than singleplex reactions, multiplex PCRs can often result in uneven amplification across the genome, with some amplicons being 100-fold more abundant than others by the end of the PCR cycles.2 This raises challenges in efficiently sequencing the genome, particularly when it comes to the low-abundance targets.
The Midnight Panel generates 1,200 bp amplicons, which is the reason the method is named “Midnight” (i.e., 1,200 = 12:00 a.m./midnight). During the development of the Midnight Panel, the team including Freed and Silander experimented with different primer sets, which generated amplicons ranging in size from 1,200 to 2,000 bp. Of the various sizes, the 1,200 bp amplicons had the most even coverage across the genome (Figure 2). The improved coverage was also maintained across a range of input amounts.2
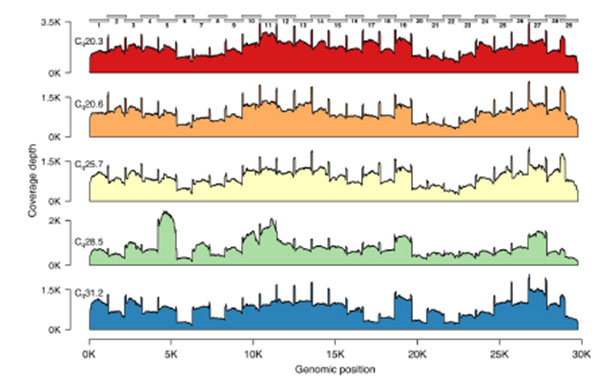
Figure 2. Genome coverage plots for patient samples varying in Cq values. The plots indicate the genome coverage for the 1,200 bp amplicon set for samples with Cq values ranging from 20.3 to 31.2. For all samples, minimum coverage exceeds 50 at all genome positions (excluding 5’ and 3’ UTR). Note that the scale of the y-axis varies between plots. The locations of the amplicons are indicated above the first plot.2
As each 1,200 bp amplicon in the Midnight Panel is amplified evenly, one needs to generate less data overall, thus saving money. The background noise and amount of redundant data that would need to be analyzed are thus minimized. Finally, the Midnight method was tested to see whether it could still adequately and reliably detect variants in cases of sample mixing. The results revealed no false positives, meaning that no variants were identified that were not in the uncontaminated original sample. This adds a notable robustness to the protocol.
While the ARTIC method was developed using a ligation-based Oxford Nanopore preparation method for the DNA library, the Midnight method uses a transposon-based Oxford Nanopore Rapid Barcode library method, which is faster and more cost-effective. Indeed, it was because the transposon-based preparation is better suited to larger amplicons that the Massey University Group looked to develop a method that generated amplicons larger than 400 bp SARS-CoV-2 ARTIC Panel amplicons.
As the SARS-CoV-2 ARTIC method was adopted by different researchers, it was quickly adapted for high-throughput sequencing, especially in countries with high numbers of infections. By using different library preparation methods, the amplicons can be made compatible with different sequencing methods. Similarly, as the Midnight Panel was used by other groups, it also was adapted to high-throughput sequencing by using a variety of library preparation methods. It has also been adapted for other long read technologies such as Pacific Biosciences.7
Controlling The Spread Of COVID-19 In New Zealand
At this stage in the pandemic, when countries like New Zealand have achieved a state where there is almost no community transmission, it was critical to develop a faster sequencing protocol to facilitate rapid responses to any new cases. As other nations get back to business as usual, it becomes even more important to act quickly to also prevent any community transmission. The Midnight Panel has been very effective in New Zealand since the sequencing data is ready within 8 to 9 hours. Such rapid turnaround means that the health authorities can use the data to make quick decisions to control and mitigate transmission event(s) from sporadic cases of COVID-19.
Indeed, such cases have cropped up and will undoubtedly continue. Where several initial transmission events have tended to stem from an index case originating from a Managed Isolation and Quarantine (MIQ) facility, the use of the rapid genome sequencing allows policy makers more concrete evidence of how the virus is being transmitted in real time and provides greater resolution around what steps can be taken to halt the spread of the disease. While the Midnight Panel is being used under these circumstances for maximal speed rather than throughput, the method can still be applied for high-throughput sequencing.
Ultimately, having more methods available will enable faster and more effective contact tracing as well as help scientists better understand the SARS-CoV-2 virus, its relation to other viruses, the mode and rate of its evolution, its geographical spread, and ramifications the mutation has to its virulence.
References
- Callaway E. The coronavirus is mutating - does it matter? Nature. 2020;585(7824):174-177.
- Freed NE, Vlkova M, Faisal MB, et al. Rapid and inexpensive whole-genome sequencing of SARS-CoV-2 using 1200 bp tiled amplicons and Oxford Nanopore Rapid Barcoding. Biol Methods Protoc. 2020;5(1):bpaa014.
- ARTIC Network. SARS-CoV-2. https://artic.network/ncov-2019. Accessed May 2021.
- Tyson JR, James P, Stoddart D, et al. Improvements to the ARTIC multiplex PCR method for SARS-CoV-2 genome sequencing using nanopore. bioRxiv. 2020.
- GISAID. https://www.gisaid.org/. Accessed May, 2021.
- Quick J, Loman NJ, Duraffour S, et al. Real-time, portable genome sequencing for Ebola surveillance. Nature. 2016;530(7589):228-232.
- Pacific Biosciences of California I. COVID-19 Sequencing Tools and Resources. https://www.pacb.com/research-focus/microbiology/covid-19-sequencing-tools-and-resources/. Accessed June, 2021.
About The Authors:
Nikki Freed is the lead technologist at Auckland Genomics at the University of Auckland, New Zealand since 2021. For six years, she was a senior lecturer at the School of Natural and Computational Sciences at Massey University, Auckland. She completed her Ph.D. at ETH Zurich in Switzerland and worked for three years at Novartis Pharmaceuticals in Basel, Switzerland. She originally hails from California.
Olin Silander has been senior lecturer in the School of Natural and Computational Sciences at Massey University, Auckland, since 2015. His work focuses on bacterial genetics and evolution. He received his Ph.D. in evolutionary biology from the University of California, San Diego, before a postdoctoral fellowship at ETH Zurich and a research fellowship at the University of Basel.
Nick Downey is currently Integrated DNA Technologies’ NGS collaborations lead. Prior to this, he served in applications support and as senior product manager. He has a Ph.D. in molecular biology along with postdoctoral experience and time as an assistant professor. He has been at IDT since 2012.
Disclaimer
The IDT products identified in this article are for research use only and not for use in diagnostic procedures. Unless otherwise agreed to in writing, IDT does not intend these products to be used in clinical applications and does not warrant their fitness or suitability for any clinical diagnostic use. Purchaser is solely responsible for all decisions regarding the use of these products and any associated regulatory or legal obligations.