
Release Specifications For Plasmid MCBs, Plasmid DNA
By BioPhorum

Plasmid release specifications are critical to the manufacture of many cell and gene therapy (CGT) products, but current guidance defining expectations for the release of plasmids as a starting material is limited. In this article, we summarize the key industry feedback, best practices, and member discussions shared in recent BioPhorum publications1,2,3 on the topic to complement ongoing efforts in the wider CGT field to advance release specifications for plasmid master cell banks (MCBs) and plasmid DNA and to de-risk the plasmid supply chain.3
Table 1 below shows an approach to platform testing. It summarizes the category of assays (i.e., identity, purity, and/or potency) and attributes and suggested methods and acceptance criteria.
Table 1: Proposed Plasmid MCB And Plasmid DNA Testing And Release Platform (extract from reference 1)
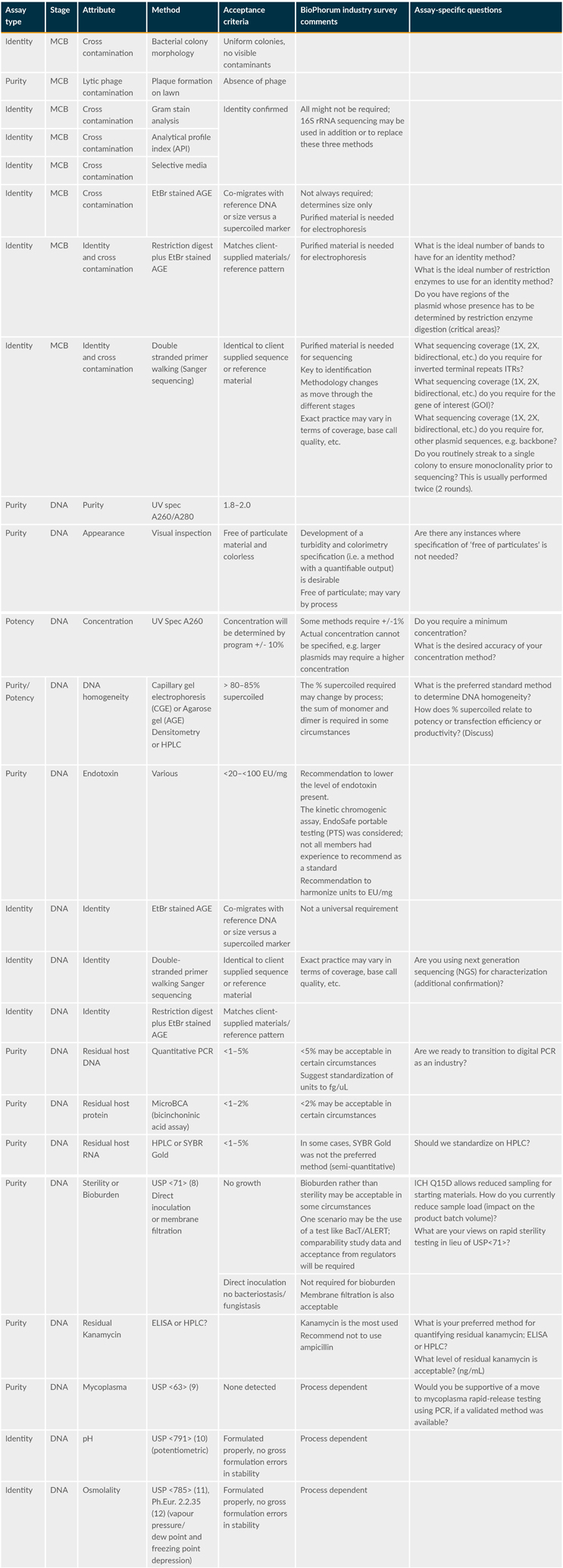
Identity Testing And Cross Contamination (Plasmid MCBs And Plasmid DNA): Multiple methods could be used to assess identity and cross-contamination of plasmid E. coli MCBs. Verification of identity is an FDA requirement for any starting material before it is used in a drug manufacturing process. Plasmid identity testing is performed upon release from the supplier and by the user before use. The supplier may use sequencing and/or restriction digest mapping as methods of identity when performing quality control release testing. In general, the supplier should perform sequencing on the entire plasmid sequence compared to a reference sequence; using restriction enzyme digestion may be helpful as a qualitative guide.
Also, the team suggests Sanger sequencing as an alternative method to confirm identity and cross-contamination. However, it is recognized that traditional Sanger sequencing may be difficult and special method adaptation is needed due to the complex challenges. These arise from the repetitive nature of sequence regions present in some plasmid DNA (i.e., repeated long terminal repeat [LTR] and inverted terminal repeat [ITR] sequence regions) and DNA templates (e.g., long stretches of poly(A) sequence regions present in mRNA-based gene therapies). Supplementary to the original paper, next-generation sequencing techniques should be used if available. These have an enhanced capacity to sequence repetitive regions on these sections of plasmid DNA or to perform a restriction digest that cleaves within the LTR or ITR sequences to demonstrate sequence integrity. The correct process will be plasmid-dependent and should be developed in partnership with sequencing data.
Appearance Testing (Plasmid DNA): The team proposes visual inspection to test plasmid DNA for appearance upon release and recommends that appearance should be colorless and free of particulate material. For added clarification, since the plasmid, in this case, is not being injected into a patient directly, consideration of microscopic particulates is not required, and such methods are excluded from the proposed release platform.
Testing For DNA Homogeneity (Plasmid DNA): The team proposes capillary gel electrophoresis (CGE), agarose gel electrophoresis (AGE), densitometry, or high-performance liquid chromatography (HPLC) as possible methods to assess DNA homogeneity upon release of plasmid DNA. Feedback received highlights that the percentage that is supercoiled may impact potency or transfection efficiency. The simpler approach for bacterial MCBs and plasmids is to record the percentage of DNA that is in a supercoiled state as determined by CGE, AGE, densitometry, or HPLC.
Testing For Residual DNA (Plasmid DNA): The team proposes quantitative PCR (qPCR) to assess residual host DNA present in plasmid DNA upon release; however, digital PCR (dPCR) technology has become widely accepted in the pharmaceutical field with many applications. Since the original publication, the team has considered dPCR for the measurement of contamination by other host cells by targeting the rRNA gene. Currently, there is no standard tool for performing the assay. Further, there is no agreed methodology to interpret the data that dPCR assays generate. Therefore, the use of dPCR to document and record residual DNA contamination has been used cautiously within the CGT field.
Testing For Residual Host RNA (Plasmid DNA): HPLC or SYBR Gold were offered as potential methods to test plasmid DNA for residual host RNA in addition to qPCR. While it is generally accepted that measurement by HPLC may provide superior accuracy, precision, and sensitivity to quantify residual host RNA, alternative methods for quantification of residual host RNA may be advantageous, particularly for plasmid suppliers that may not have HPLC capability. Fluorescent gel staining methods may be suitable as an alternative to HPLC.
Testing For Sterility Or Bioburden (Plasmid DNA): The team recommends testing by USP <71> direct inoculation or membrane filtration. Feedback highlights that low bioburden rather than sterility may be acceptable in some circumstances, and the team acknowledges that unique sterility or bioburden requirements for plasmids may be defined through a risk-based approach, based on where and how the plasmids are added to the user’s processes.
Testing For Residual Kanamycin (Plasmid DNA): Plasmids containing kanamycin resistance genes (e.g., nptII) and other antibiotic-resistant genes are not uncommon in commercial production and allow for a practical cell selection process of target cells over plasmid-free cells. Kanamycin is an aminoglycoside that binds the 30S ribosomal subunit and causes mistranslation in the bacterium. Often used at working concentrations of 50 to 100 μg/mL in cell selection, toxicity to human health and non-resistant cell cultures necessitate some level of control and evaluation in raw material plasmid products.
Elemental, Extractable, And Leachable Impurities: On the user side of transduction, the number and extent of the elemental, extractable, and leachable impurities associated with the raw material plasmid and its closure can make process development challenging. However, it may be prudent to consider the overarching critical quality attributes. In the scope of upstream manufacturing, it may be practical to assess the integrity of the plasmid material (e.g., supercoiling, percentage of nicking, identity, integrity) as an orthogonal measure of the effect of any of these impurities exerted on the material.
Demonstrating Comparability Due To Changes In The Plasmid Manufacturing Process: Considerations from the International Council for Harmonization (ICH) Q5E Comparability of biotechnological/biological products may be applied to demonstrate that changes to the plasmid manufacturing process do not adversely impact the quality, safety, and efficacy of the plasmids. The rigor around evaluating comparability should be on par with the treatment modality the plasmids are used in and in proximity to the final drug product/patient, phase of development, and associated manufacturing/clinical experience and history.
Stability Studies: Stability study design, requirements, and interpretation of results for plasmids can be acquired directly from ICH quality guidelines, including Q1A (R2) Stability testing of new drug substances and products, Q1D Bracketing and matrixing designs for stability testing of new drug substances and products, and Q1E Evaluation of stability data. While these ICH guidelines are recommendations intended for drug substances and drug products, the concepts and suggestions can be applied to plasmids used as starting materials in CGT.
This article is a summary of BioPhorum publications1,2,3 on the topic. To read more, check out the references below, including our most recent publication, Discussion on plasmids to establish release specifications using a risk-based approach to manage supply.
Moving Forward
Since the original BioPhorum paper1 was published in 2020, the EMA issued specific guidance in 2021 that plasmids should be made using GMP conditions.4 The degree to which a supplier can meet these requirements and provide documentary evidence to support them is central to the use of plasmids in the CGT industry.
In 2020, the U.S. Pharmacopoeia formed an expert panel to draft a new chapter to document and standardize plasmid release specifications where plasmid DNA will be used as a starting material for the manufacture of CGTs. Applying a common standard will enable communication and cooperation across the pharmaceutical industry and simplify communication with regulators. We welcome the publication of this common standard.
BioPhorum will likely revisit the topic as further data is released, for example, on the use of next-generation sequencing, new processing developments, or expansion in knowledge of extractables and leachables.
References
- Raw Materials: Cell and gene therapy critical starting material: a discussion to help establish release specifications for plasmids and the bacterial master cell banks used to produce them, BioPhorum, 2020
- Raw Materials: Perspectives on raw and starting materials risk assessment for cell and gene therapy (CGT) processes, BioPhorum, 2020
- Discussion on plasmids to establish release specifications using a risk-based approach to manage supply, BioPhorum, 2022
- European Medicines Agency EMA/246400/2021 Questions and answers on the principles of GMP for the manufacturing of starting materials of biological origin used to transfer genetic material for the manufacturing of ATMPs, (2021)