
Regulatory Pathways — With Real-Life Examples — For Successful CGT Clinical Trials
By Jessica Cordes, senior consultant, Clinical Excellence GmbH

Cell and gene therapies (CGTs) have been established in the treatment landscape across oncology, rare diseases, and inherited disorders. Yet the clinical trials required to bring these therapies to market present significant operational and regulatory complexity. For small biotech companies transitioning into the clinical phase, regulatory strategy is not a post-approval consideration. It is a critical enabler of clinical trial feasibility, speed, and eventual market access.
In this article, I explore the most effective regulatory pathways used to support CGT development and highlight two real-life examples that illustrate how small and midsize biotechs can optimize clinical trial design, site and manufacturing readiness, and regulatory alignment. By integrating regulatory expectations into early development planning, companies can reduce risk, maximize resource efficiency, and position themselves for sustainable success.
Regulatory Frameworks For Cell And Gene Therapies
The regulatory landscape for CGTs has evolved rapidly in recent years. In the United States, the FDA has established several pathways to support innovative therapies, including Breakthrough Therapy designation, Regenerative Medicine Advanced Therapy (RMAT) designation, Fast Track, and Priority Review. In the European Union, the EMA offers similar accelerated pathways such as PRIME (PRIority MEdicines), accelerated assessment, and the orphan medicinal product designation. Japan has also created an expedited review framework for regenerative and cellular therapies under the Sakigake designation.
These pathways aim to address the unique challenges CGTs face with small patient populations, novel endpoints, complex manufacturing, and long-term follow-up requirements. For small sponsors, understanding how to combine these designations and align with regulatory expectations early can unlock meaningful efficiencies in time and resources.
Enabling Success Through Early Regulatory Engagement
The most successful CGT clinical trials typically share three core regulatory features:
- Early engagement with regulators to align clinical trial design and risk mitigation strategies
- Use of special designations to gain regulatory support and priority interactions
- Early operational readiness to meet CMC, safety, and quality expectations from first-in-human (FIH) through commercialization
These principles are exemplified in two therapies that reached approval through distinct but instructive pathways.
Case Study 1: Luxturna – Gene Therapy For RPE65-Mediated Retinal Dystrophy
Luxturna (voretigene neparvovec) is an adeno-associated virus (AAV)-based gene therapy approved for patients with confirmed biallelic RPE65 mutation-associated retinal dystrophy. The therapy delivers a functional copy of the RPE65 gene directly to retinal cells.
Key regulatory milestones include:
- FDA Breakthrough Therapy designation
- EMA Priority Medicines (PRIME) status
- Orphan Drug designation (U.S. and EU)
- Novel endpoint development with multi-luminance mobility testing (MLMT)
Trial sponsor Spark Therapeutics engaged early with both the FDA and EMA to validate their novel endpoint, confirm manufacturing controls, and agree on durability follow-up requirements. Their pre-IND interactions focused heavily on endpoint justification and vector delivery methodology.
From a clinical trial execution standpoint, sites were trained to conduct sub-retinal injections and manage cryogenic supply chains. The regulatory strategy integrated manufacturing scale-up and safety monitoring from the outset, enabling the therapy to progress from Phase 1/2 to approval in under five years.
Case Study 2: Yescarta – CAR-T Therapy For Relapsed/Refractory LBCL
Axicabtagene ciloleucel (Yescarta) is a CD19-directed autologous CAR T cell therapy approved for the treatment of adult patients with relapsed or refractory large B-cell lymphoma.
Key regulatory milestones:
- FDA Breakthrough Therapy designation
- Priority Review designation and accelerated approval
- EMA accelerated assessment
In the pivotal ZUMA-1 clinical trial, Yescarta demonstrated strong efficacy in a high unmet-need population. The regulatory success of this program relied on:
- Close collaboration with the FDA on safety monitoring (e.g., cytokine release syndrome and neurotoxicity)
- Clear demonstration of manufacturing robustness for autologous products
- Realistic long-term follow-up plans embedded in the initial clinical protocol
Site preparation and cross-functional coordination were central to execution. Apheresis units had to manage cryogenic chain-of-identity processes, while clinical trial sites required dedicated training on toxicity grading, dose preparation, and patient monitoring. The regulatory team maintained ongoing dialogue with both the FDA and EMA on comparability assessments and real-world data planning.

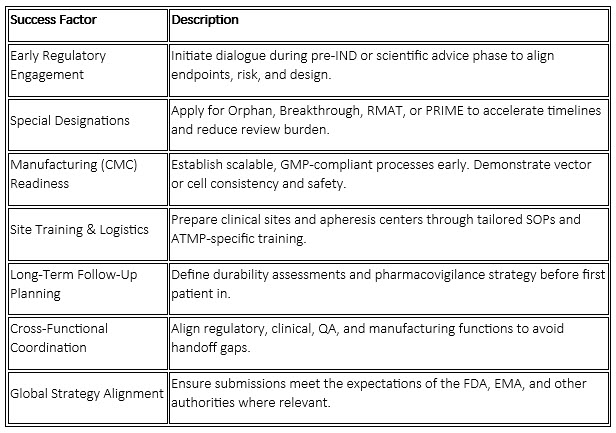
Success Factors And Considerations For Small Biotech Sponsors
For small and emerging biotech sponsors, regulatory success is not only about designations but also readiness and integration:
Regulatory agencies are moving beyond traditional product approval pathways by encouraging more innovative and flexible approaches in CGT clinical trial design. This includes the adoption of adaptive clinical trial designs, which allow for modifications to clinical trial protocols based on interim data without compromising the integrity or validity of the clinical trial. Such designs can accelerate development timelines by enabling sponsors to adjust sample sizes, endpoints, or even patient populations as new information emerges during the clinical trial.
Beyond traditional data from randomized controlled trials, real-world evidence (RWE) is becoming increasingly important in regulatory submissions for CGTs. RWE encompasses data gathered from sources such as electronic health records, patient registries, insurance claims, and even wearable devices or mobile health applications. This type of evidence reflects how therapies perform in everyday clinical practice, capturing a wider range of patient experiences, including those from populations who may not typically participate in clinical trials due to age, comorbidities, or geographic location.
Regulators like the FDA and EMA are encouraging the use of RWE to complement clinical trial data, as it can help demonstrate a therapy’s effectiveness and safety in real-world settings. Incorporating RWE allows for a more holistic understanding of a product’s risk-benefit profile, supports post-market surveillance, and can even inform label expansions or new indications. In CGT, where patient populations are often small and diseases are rare, RWE is particularly valuable for assessing long-term outcomes, durability of response, and the occurrence of rare adverse events. By leveraging RWE, sponsors can provide regulators with more robust and relevant data, ultimately facilitating faster and more informed decision-making throughout the therapy’s life cycle.
For small biotech companies operating with limited resources and tighter budgets, adopting patient-centric approaches may initially seem challenging. However, even with these constraints, it is possible to meaningfully involve patients in the clinical development process by focusing on practical, scalable strategies.
- Practical Patient-Reported Outcomes (PROs): Small companies can use simple, validated questionnaires or digital surveys to gather patients’ experiences regarding symptoms, quality of life, and treatment satisfaction. Leveraging existing tools reduces development time and cost, while still providing valuable, actionable insights that go beyond traditional clinical endpoints.
- Collaboration with Patient Advocacy Groups: Building relationships with advocacy organizations doesn’t require a large budget. Small biotechs can invite representatives to participate in protocol review meetings or solicit feedback via virtual focus groups. Their input can help ensure trial designs address real-world needs and enhance patient recruitment with minimal financial investment.
- Enhancing Accessibility and Convenience: While comprehensive decentralized trial platforms may be costly, small companies can still improve accessibility by offering flexible visit windows, using telehealth for select follow-up appointments, or partnering with local clinics for lab work. Providing clear instructions and support for remote data entry, such as simple mobile apps or telephone check-ins, can also minimize participant burden without significant infrastructure costs.
- Simple Feedback Loops and Communication: Establishing ongoing feedback can be as straightforward as periodic phone surveys or short online questionnaires. Assigning a dedicated contact person for participant questions and providing regular updates (e.g., newsletters or email summaries) helps build trust and keeps patients engaged, even without elaborate platforms.
- Promoting Diversity and Inclusion: Small companies can collaborate with local clinics, community health centers, or advocacy groups to reach underrepresented populations. Tailoring recruitment materials to different languages and cultural contexts and ensuring eligibility criteria are inclusive can broaden trial participation without requiring substantial additional resources.
By prioritizing these manageable patient-focused strategies, small biotech companies can create clinical trials that are meaningful, inclusive, and attractive to participants. Patient centricity does not necessarily require large budgets; instead, it can be achieved through thoughtful planning, leveraging community partnerships, and maintaining open lines of communication with clinical trial participants. These efforts not only improve recruitment and retention but also ensure the data generated are relevant and valuable for regulatory submissions and future clinical care.
Ultimately, patient-centric strategies not only improve recruitment, retention, and overall satisfaction but also generate richer, more relevant data. This helps ensure that the therapies under investigation address real patient needs, which is critical for successful regulatory approval and meaningful advancements in clinical care.
Together, these regulatory trends aim to make CGT development more efficient, responsive to patient needs, and better aligned with real-world clinical practice. Sponsors who proactively adopt these approaches can facilitate regulatory review, improve data quality, and ultimately bring transformative therapies to patients more rapidly.
By combining clear designation strategies, robust process documentation, and site-level execution support, sponsors can de-risk their clinical journey and bring transformative therapies to patients faster. With the right support, even small companies can navigate complex global regulatory pathways and emerge with high-quality data, regulator confidence, and sustainable development strategies.
About The Expert:
 Jessica Cordes started her clinical operations career in 2009, working at various companies including Big Pharma and several small to midsize biotech companies. She gained extensive experience on different levels from country study management to global study management and, since 2018, leadership in clinical operations. During her time at Medigene and Immatics, she structured the clinical operations department, built cohesive global teams, and implemented GCP and ATMP-compliant processes. For more than 12 years, she has been working in oncology clinical trials (including hemato-oncology as well as solid tumors) and with ATMPs since 2018. Since 2023, she has been working as an independent consultant and trainer, supporting small companies in building their clinical operations group and setting up their clinical trials for success. She provides a GCP refresher course via her Clinical Excellence Training Academy.
Jessica Cordes started her clinical operations career in 2009, working at various companies including Big Pharma and several small to midsize biotech companies. She gained extensive experience on different levels from country study management to global study management and, since 2018, leadership in clinical operations. During her time at Medigene and Immatics, she structured the clinical operations department, built cohesive global teams, and implemented GCP and ATMP-compliant processes. For more than 12 years, she has been working in oncology clinical trials (including hemato-oncology as well as solid tumors) and with ATMPs since 2018. Since 2023, she has been working as an independent consultant and trainer, supporting small companies in building their clinical operations group and setting up their clinical trials for success. She provides a GCP refresher course via her Clinical Excellence Training Academy.