
Managing The Presence Of Visible Particulates In Cell Therapies
By BioPhorum

Cell therapies (CTs) face unique challenges in visible particulate control and detection compared to other injectables. These include difficult-to-inspect formulations and containers, inherent cell-related particulates that limit the detection of other particles, and terminal sterilizing filtration that is not applicable due to cell size and formulation needs. For small-batch volumes, especially in autologous therapies, rejecting units with particle defects significantly impacts supply continuity.
Regulatory guidance and health authority expectations are also not aligned with the unique characteristics of CTs, posing challenges for sponsors in meeting particulate specifications. The presence of visible particles in any biological drug product (DP) is a significant concern for regulatory authorities, as it can lead to product rejection and recalls, clinical complications, and compromised patient safety.
This article provides a holistic approach to developing a comprehensive particulate control strategy for CTs, focusing on particle characterization, detection, and manufacturing controls.
Current Guidance
The requirements in current guidance are primarily designed for parenteral products, including those that are difficult to inspect. Given the unique characteristics of cell-based products, implementing these requirements is typically infeasible.
Figure 1 summarizes the regulatory challenges associated with visible particle control for CT products. Each hexagon represents a unique characterization of advanced therapy medicinal products. The orange text represents associated current guidance. The white text highlights specific points in the guidance that we believe are not practical for CT products.
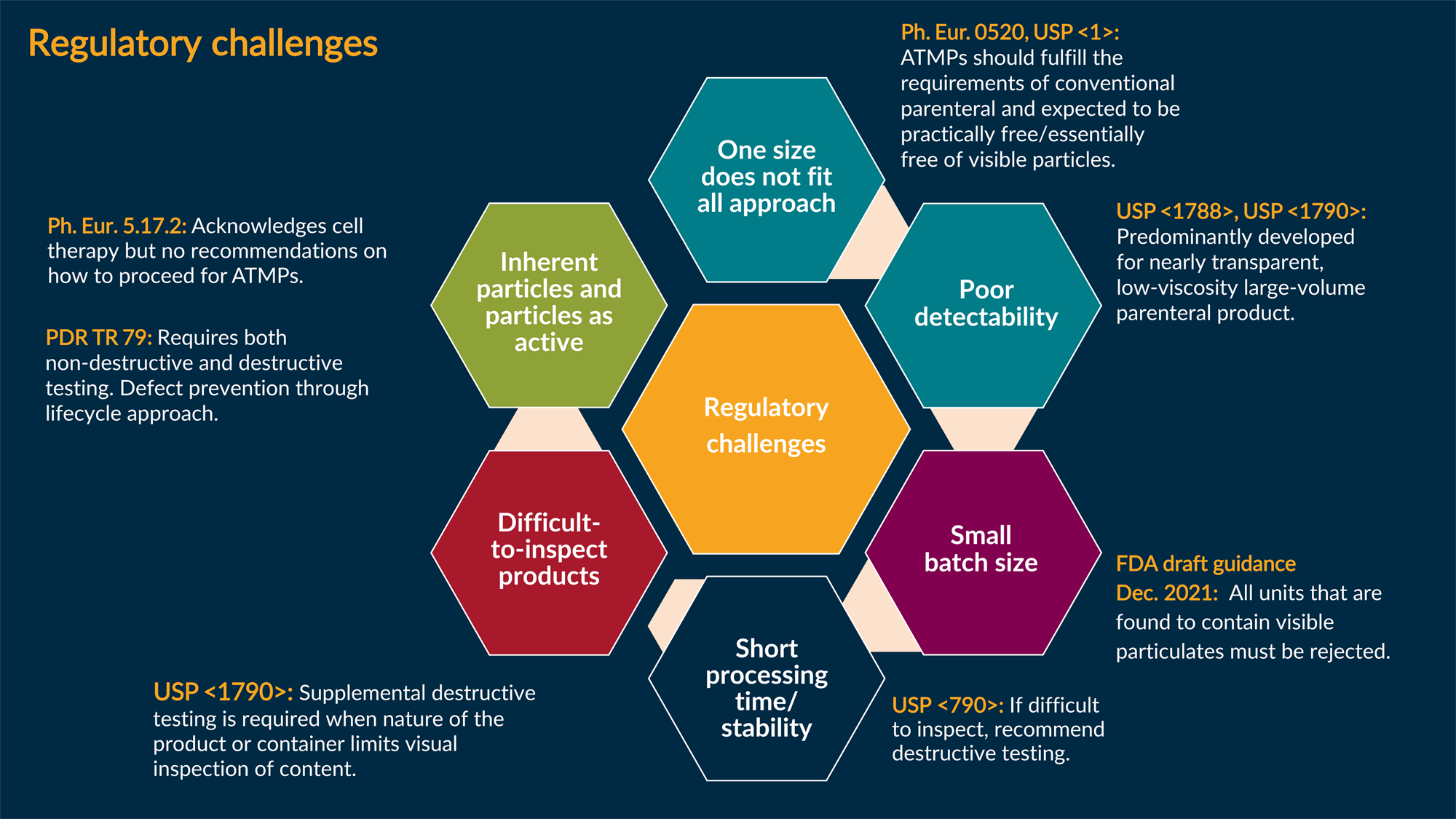
Figure 1: Regulatory challenges associated with visible particle control for cell therapy products.
Key: ATMP — Advanced therapy medicinal product.
Click on image to enlarge.
Challenges And Limitations With Visual Inspection
For visual inspection (VI), CT products have unique challenges and considerations compared to other modalities. An overview of these challenges is shown in Figure 2.
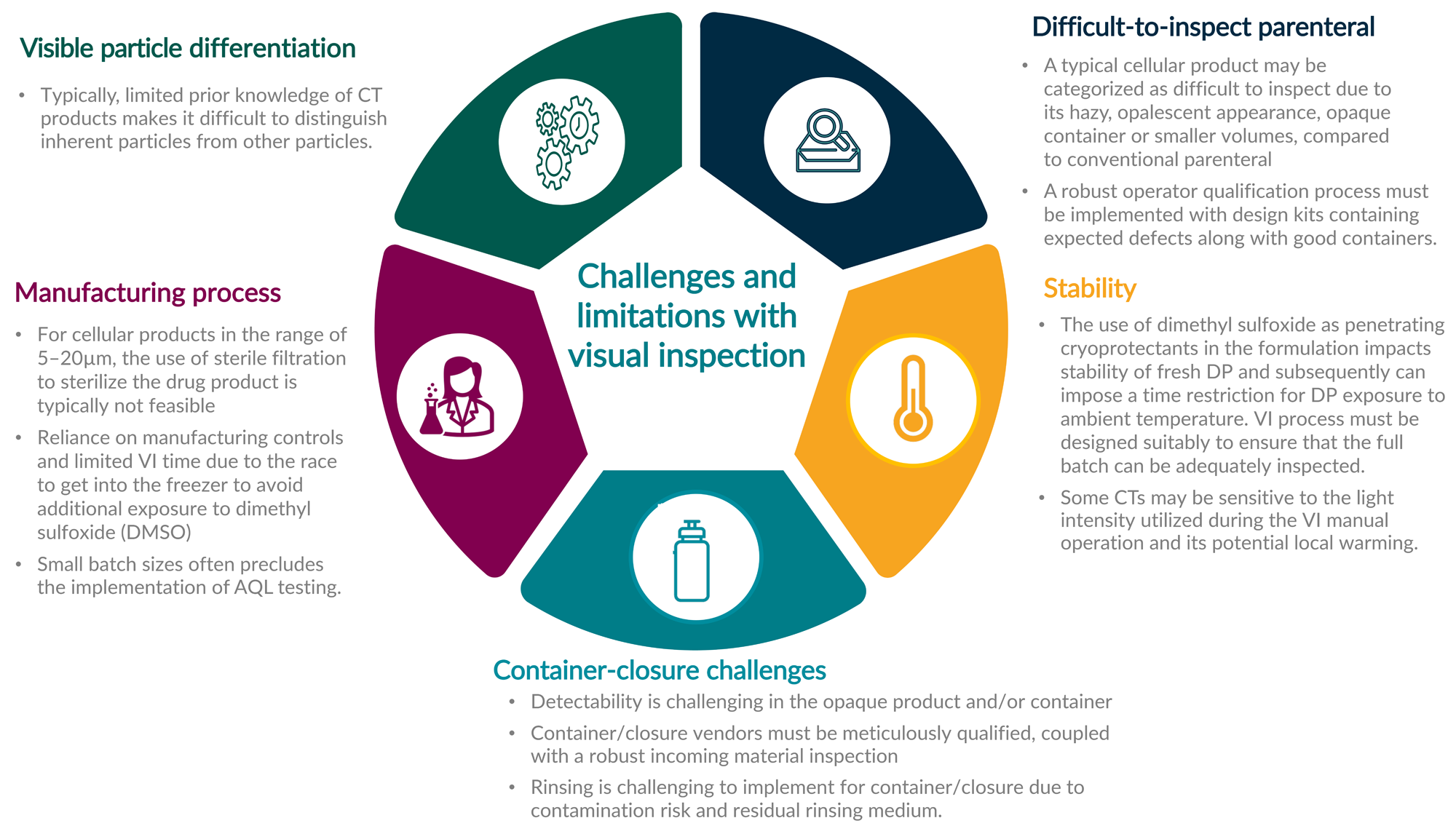
Figure 2: A summary of challenges associated with visual inspection for cell therapies.
Key: AQL — Acceptable quality level.
Click on image to enlarge.
Potential Impact
A proactive approach is needed early in development to understand, characterize, and detect particles. For CTs, the risk of particulates in DP cannot be eliminated; it can only be mitigated through controls. Often, destructive testing may not be feasible; therefore, building a knowledge library is key to helping CT manufacturers assess common particles without destructive testing. This will need to be a phase-appropriate approach.
Potential Impact Of Particulate On Patients
When setting an appropriate specification for particulates, the primary goal is to ensure patient safety, and there is substantial literature evaluating the potential risks to patients. The amount of risk depends on several factors including particle classification (inherent, intrinsic, or extrinsic), patient factors, route of administration, and particle characteristics. We recommend that developers/manufacturers use a risk-based approach to assess the appropriate specifications. Risk assessments should consider particle classification, particle characteristics, patient-specific factors, and route of administration to ensure an appropriate benefit-risk evaluation.
To support decision-making when visible particles are detected, we propose a risk-based decision tree that guides particle classification, characterization, investigation, product disposition, and patient benefit-risk assessment (see Figure 3).
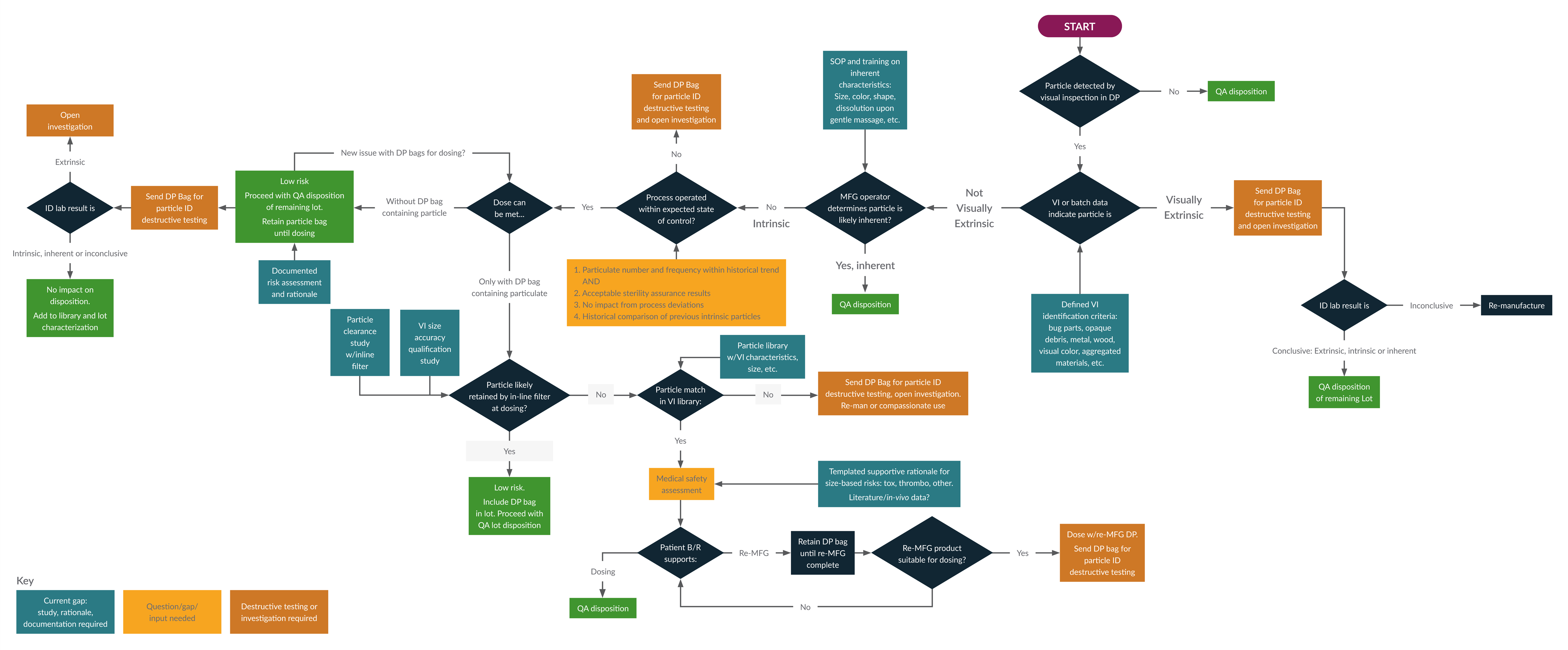
Figure 3: Decision tree to understand the risk of a particulate. Click on image to enlarge.
Particle Characteristics
The specific characteristics of particulates identified in a CT are critical in assessing the risk to patient safety. Factors such as particle size, quantity, and reactivity may significantly increase safety risks. In gene therapy products, large particulates can be removed via filtration; however, the lack of terminal filtration in most CT products increases the risk of visible particles that may clog delivery systems or pose a thromboembolic risk, particularly if present in high quantities.
Root Causes And Characterization
The likelihood of particulates in CT DPs is higher than in other sterile-filtered products because sterile filtration cannot be applied; therefore, particulate control must be built in. One of the first steps to achieve this is to understand the process and contributors to particulate matter from different consumables and product-contact surfaces.
Determining Potential Sources In The Process
Eliminating all particulates from the final DP of any CT therapy has practical limitations; however, the particulate load of the final therapy must be minimized and characterized. The first step is to understand and identify the potential sources of particulates in the manufacturing process.
For manufacturing processes, we recommend gathering process knowledge of all potential particulate sources through an Ishikawa diagram (see Figure 4) to evaluate the associated methods, machinery, environment, materials, and manpower.
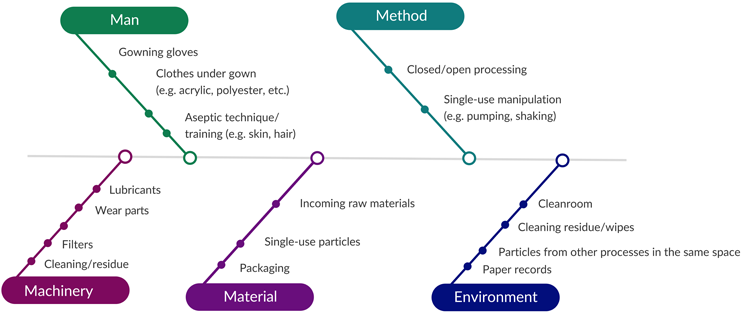
Figure 4: Ishikawa diagram showing potential sources of visible particles
Techniques For Identifying And Characterizing Particulates
With the identification of potential sources and root causes of intrinsic or extrinsic contamination completed, the deductive work to identify and characterize the particulate matter can be accelerated to determine which analytical assays are suitable. Historically, regulatory agencies such as USP and Ph. Eur. have established general chapters on VI and, specifically, particulate matter regarding liquid injectables that are potentially relevant to visible particles in CTs.
An idealized process flowchart for characterizing particulate/foreign matter in any pharmaceutical drug substance, DP, raw material, single-use systems (SUS), process intermediate, or equipment is shown in Figure 5.

Figure 5: Typical workflow for foreign matter/particulate matter evaluation
In general, the best approach for identifying and characterizing visible particulate matter should employ as many of the above techniques as feasible. These should consider the limitations of each assay to the visible particulate type specific to the process-specific drug substance or DP and ensure it is within the suitable size range and chemical composition (e.g., hydrophilic or hydrophobic), with appropriate dispersity.
Common Particulates
You should develop a material process map to trace potential visible particles to the materials in equipment, SUS, and components, as well as the ancillary tools used in the process steps up to and including the final fill of DP. Along with this map, it is critical to understand the material of construction of the particle contamination found during manufacturing. A compilation of particles identified during manufacturing via deviations and the process particle traceability map can then be made. This compilation will yield a list of particle materials of concern that are likely and potentially observed throughout the manufacturing process.
Risks From Visible Particles Present In Single-Use Bioprocessing Equipment And Final Drug Product Containers
Multi-use bioprocessing equipment (e.g., stainless steel) and some final DP containers (e.g., glass vials) are cleaned with chemicals and rinsed with high-purity water before sterilization. One objective of these cleaning processes is to reduce the level of visible particles present on the interior (fluid-contacting) surfaces. However, presterilized single-use bioprocessing equipment and many types of final DP containers are often not cleaned or rinsed before implementation in the manufacture of CTs.
Control Strategy
Here, we discuss establishing a particle control strategy, methods for controlling the introduction of particulates into DP, and guidance on responding when a particulate is identified in DP.
Process Characterization Flowchart
Characterization of particulates from SUS may be conducted using a plethora of analytical techniques. A process-flow analysis can further support identification and characterization of particulates from the full process. Figure 6 details the basic steps of process characterization from the perspective of particulate characterization in a CT process.
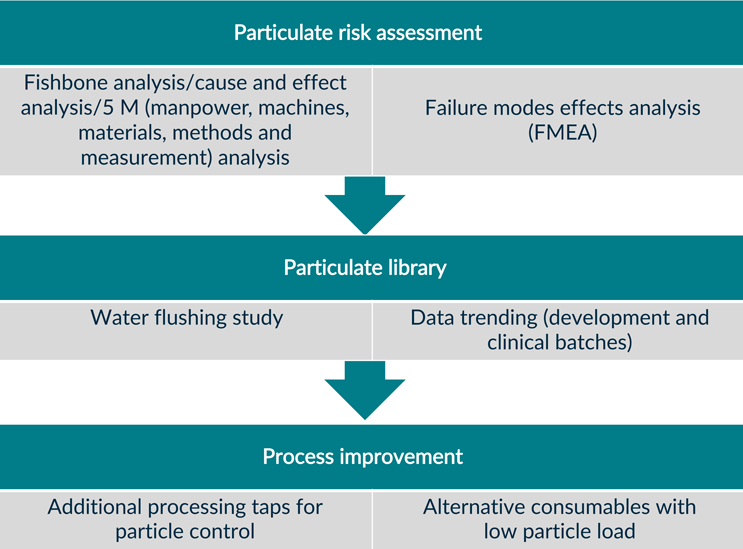
Figure 6: Process characterization flowchart for particulates in cell therapies
Since visible particulates can be intrinsic, inherent, or extrinsic, characterizing each of these particulates follows a slightly different path (see Figure 7 for a process flow for intrinsic particulates).
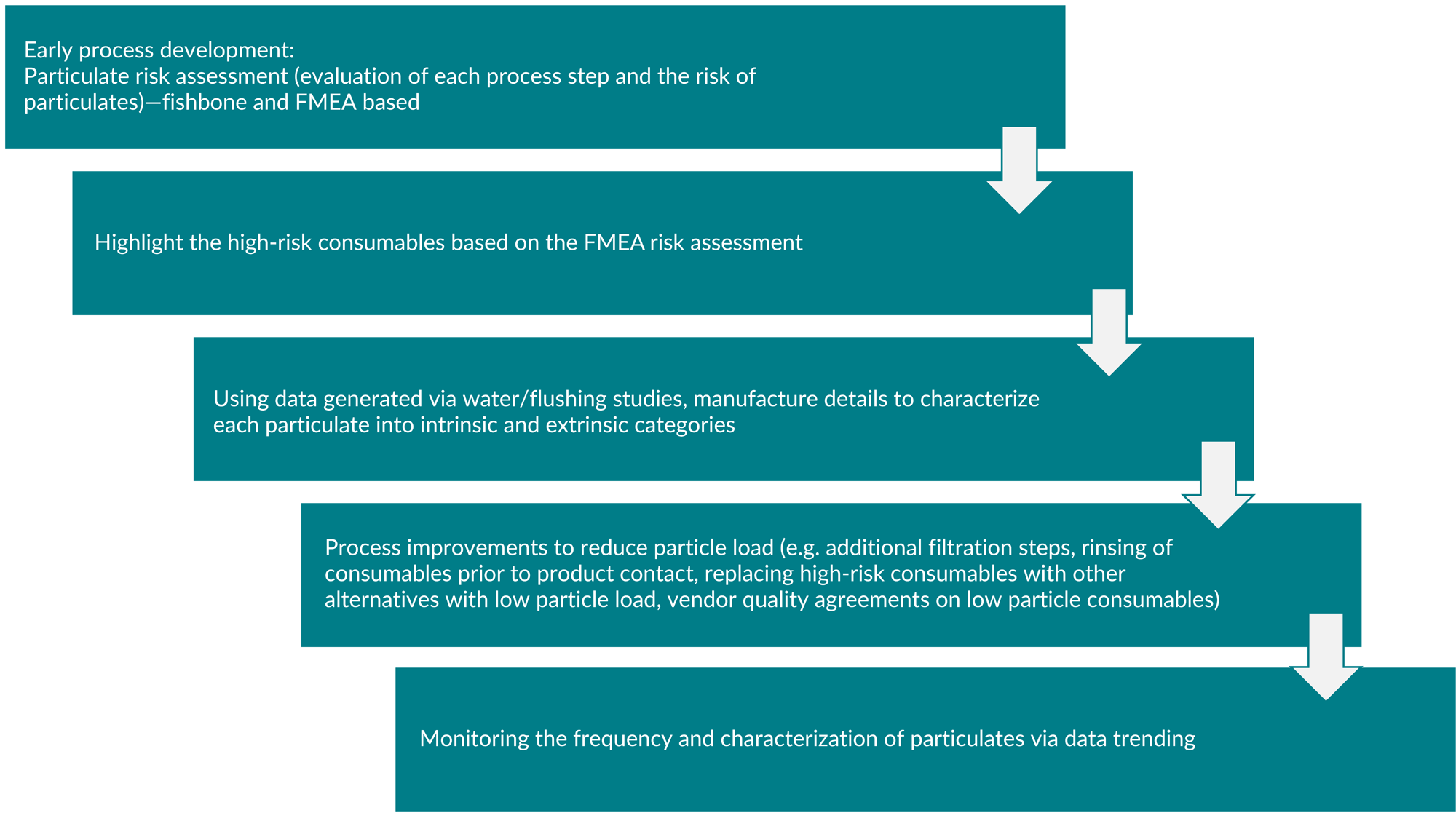
Figure 7: Process characterization flowchart for intrinsic particulates in cell therapies.
Key: FMEA — Failure mode effects analysis.
Click on image to enlarge.
A phase-appropriate approach is required to establish the particle control strategy. The timeline for characterization should begin with the early process development phases; a risk assessment for particulate introduction can be evaluated in parallel. The risk assessment can be initiated with a fishbone analysis and a cause-and-effect-based risk assessment, followed by a failure mode and effects analysis-based risk assessment.
Water Flush
Assuming that appropriate aseptic and manufacturing controls are implemented in a CT manufacturing process, the most likely detected visible particulates can be considered intrinsic or inherent to the manufacturing process. Single-use consumables are the most likely source of intrinsic particles, which can be extensively characterized through water-flushing studies, also known as rinsing studies or water-runs. The data from water-flushing studies is most effective when done as a combination of:
- single-use consumable flushing study
- end-to-end manufacturing process flushing study.
Vendor Controls
The best way to reduce particle events in DP is to prevent their entry. Across the SUS supplier base, there is no standardization for visible particulates or VI controls. We recommend establishing internal particulate criteria in user requirement specifications or other specifications before engaging with suppliers. This will ensure the sponsor and the supplier are aligned on particulate requirements. As part of the supplier management process, we recommend performing activities such as on-site technical due diligence, audits of the manufacturing sites, and establishing a quality agreement and requirements for particulates.
Material Controls
For higher-risk materials, we recommend performing appropriate material qualification with respect to applicable particulate limits or specifications. It is also important to understand lot-to-lot variability and differences between suppliers for comparable items. For particulate testing, we recommend applying compendial methods for testing and inspection (USP <788> and USP <790>).
Process Controls
The primary contributors of potential particulates could be identified by performing water or buffer simulation with the manufacturing process and evaluating in-process samples by using VI and/or imaging/microscopy. We recommend standardizing this “water-run” procedure so that the process steps, VI, microscopy, and testing are all representative of the manufacturing process and are performed under protocol. Care should be taken to design a study in which the contribution of individual components can be assessed and risk-based identification testing is performed, given the expected large volume of samples and recovered particles.
Environmental And Facility Control (Aseptic Controls)
In general, controls designed to prevent microbiological contamination also prevent many potential sources of extrinsic particulate contamination. One advantage of a robust aseptic control strategy is that it can serve as an effective prevention measure for both visible and subvisible particulates. Each manufacturing site should have a robust contamination control strategy, with defined elements using a risk-based approach. Elements that may exist within a contamination control strategy include:
- sterile filtration of prepared reagents
- personnel gowning, hygiene, and training
- facility design
- process design
- facility and equipment disinfection
- material selection
- environmental monitoring.
Visual Inspection Qualifications
Cell-based therapies are particularly challenging due to batch size (e.g., low-volume, single-dose), inherent particles, complex process, and DP sterile filtration limitations. Many organizations use a 100% lightbox inspection followed by an acceptable quality level inspection, or a second-operation inspection with light intensity of 2,000 to 3,750 lux. Also, testing times of 5, 10, and 20 seconds are commonly used for VI testing.
Appearance Testing
EudraLex Volume 4 offers an alternative control strategy for cell-based CTs. It says: “As cells in suspension are not clear solutions, it is acceptable to replace the particulate matter test by an appearance test (e.g., color), provided that alternative measures are put in place, such as controls of particles from materials (e.g., filtration of raw material solutions) and equipment used during manufacturing, or the verification of the ability of the manufacturing process to produce low particle products with simulated samples (without cells).”
Sterility Testing
Extrinsic particulate contamination can either carry live microbes or be sterile, depending on the source of contamination. Sterility testing is a basic requirement and can ensure the safety of any CT.
There are limitations to the sterility test, including difficulty detecting low inoculum levels of contamination and the fact that representative samples are only a small portion of the batch. Due to the limitations of this destructive assay, sterility testing should not be considered as a primary control for particulate contamination. For example, a particle contaminant from personnel working in the area (e.g., hair) would likely be detected by a positive sterility result; however, particle contamination from components that have been sterilized before use would not be detected by sterility testing.
Particle ID Testing
In addition to quantifying particles, it is important to identify and characterize observed particles to understand their type, source, and composition. Several commonly used particle characterization methods can be successfully applied to CTs, including scanning electron microscopy, micro-Fourier transform infrared spectroscopy, magnetic resonance spectroscopy, energy-dispersive X-ray spectrometry, and flow imaging microscopy.
Once particles have been identified, their likely source can be determined and appropriate controls implemented to eliminate future particulates from that source. See Figure 8 for a model of the life cycle management of particulates.
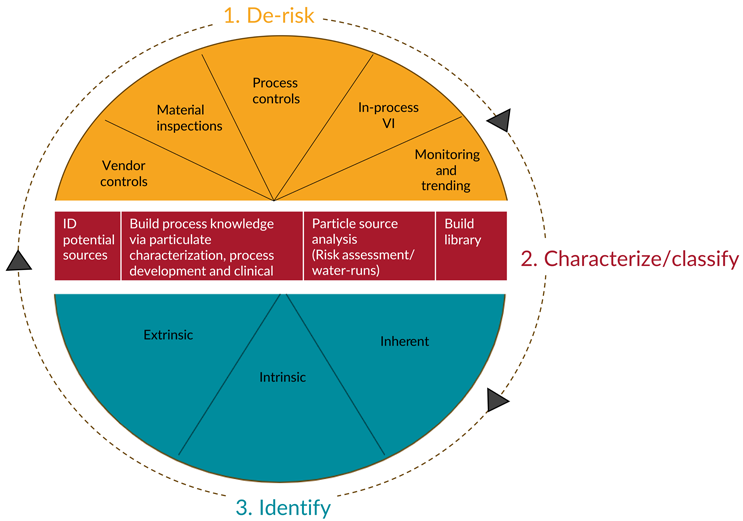
Figure 8: Proposed model for the life cycle management of particulates
Summary
CTs pose unique challenges for particulate control and detection due to the inherent cell-related particulates and other particles. Regulatory guidance is not aligned with the unique characteristics of CTs, posing challenges for meeting visible particulate specifications.
We recommend a nuanced approach to administering products with low levels of visible intrinsic particles, given that the therapeutic benefits of CT DPs often outweigh potential risks. Moreover, recognizing the characteristics of the particles (including size, type, and other factors) can further inform risk assessments. With critically ill patients relying on these therapies, the focus should pivot toward a comprehensive risk evaluation that integrates patient-specific factors, the administered volume, and the route of administration. The goal should remain particle-free but the presence of a limited number of visible intrinsic particles should not automatically preclude the release and use of these products.
We advocate for a science- and risk-based approach when assessing visible particulates in CT products. While the goal should remain to minimize particulate presence wherever possible, a limited number of well-characterized intrinsic particles should not automatically preclude product release when patient benefit outweighs the associated risk.
Manufacturers, clinicians and regulatory bodies must collaborate to establish a framework that balances safety and accessibility, ensuring that advancements in CT continue to positively impact patient outcomes. This should include greater regulatory alignment and harmonized guidance that reflects the realities of CT manufacturing, where inherent particles, limited batch sizes, difficult-to-inspect products, and the practical limitations of traditional particulate control strategies require a risk-based approach rather than conventional parenteral assumptions.
This article summarizes the main points from a recent BioPhorum paper on this topic. To learn more, check out the full paper, A holistic and strategic approach to characterize, identify and de-risk the presence of visible particulates in cell therapies. It includes appendices for key guidance, a checklist of actionable recommendations, and a summary of warning letters.