
Leveraging Human Genetics To Advance Cell Therapies For Treatment Of Blood Cancers
By Abhinav Dhall, Carolyn Wills, Nathan Jorgensen, Tirtha Chakraborty, and John Lydeard at Vor Biopharma

Developing novel therapies is a challenging endeavor as less than 10% of the drugs (chemical, biological, biotech or radiopharmaceutical) that enter Phase 1 clinical trials successfully advance to gain FDA approval (1). Nearly three-quarters of novel therapies fail during development as a result of a poor safety or efficacy profile. Improved strategies to de-risk drug targets are needed to increase the efficiency of drug development. Human genetic data are a powerful asset to identify and de-risk new therapeutic targets. A recent analysis of approximately 23,000 drugs in various stages of the pipeline revealed that drugs with supporting human genetic data are twice as likely to be approved at the end of their clinical development (2-4).
Since the early 2000s, the lowered cost, increased efficiency, and higher throughput of DNA sequencing technology has enabled vast amounts of human genome data to be generated. Scientists are using these data to identify millions of genetic variants within the human genome and characterize their impact on health and diseases (5). One early application of using human sequencing data for drug development occurred with the discovery of genetic variants in the proprotein convertase subtilisin/kexin type 9 (PCSK9) gene (6). Single nucleotide variants resulting in loss of function (LoF) in PCSK9 were found to correlate with lower cholesterol levels. (7) This informed the development of novel therapeutics, including the FDA approved therapy evolocumab (Repatha®), a monoclonal antibody inhibitor of PCSK9 designed to reduce high cholesterol in individuals at risk for coronary heart disease (CHD). This, among other examples, (8) illustrate the value of leveraging human genetic data in the development of life-saving therapies. (9)
In the last decade, the number of cell and gene therapies in development has increased exponentially (10). These therapeutics have different development challenges and risks compared to small molecules or antibodies. Specifically, gene ablation and other genetic manipulations require understanding the dispensability of the underlying target to assess the risks involved with gene editing. Using human genetic databases to identify loss of function variants in healthy individuals provides critical information on the dispensability of a gene editing target and therefore on the risk profile of the gene therapy. Herein, we describe a schema for evaluating a target and assessing the risk of its genetic manipulation using LoF variant analysis to assess gene dispensability. This schema has the potential to help cell and gene-editing drug developers prioritize programs and potentially avoid costly program failures. We first provide an overview of the genetic variation observed in humans and then highlight the critical role human genetic data played in the development of a novel gene-knockout based cellular therapy for treatment of acute myeloid leukemia (AML), starting with CD33 (11-13).
Human Genetic Variation
The genetic sequences between any two individuals differ by approximately one-tenth of a percent (0.1%), which means that one base pair out of every 1,000 is estimated to be different between any two people. The difference in DNA sequence within a population is termed genetic variation; it can arise from many types of genetic differences such as single nucleotide substitutions or nucleotide insertions, deletions, and duplications. Although genetic variation is critical for helping organisms adapt to their environment, variation in certain areas of the genome can negatively impact health.
As described in Havrilla et al., the impact of genetic variation on human health can be understood through the analogy of survival bias which was famously used by Abraham Wald during World War II (14-16). Survival bias is the logic of concentrating on the people or things that pass some selective pressure while overlooking those that do not. Wald and the Statistical Research Group examined patterns of bullet holes on planes that safely returned from battle and deduced that plane armor should be reinforced where bullet holes were not observed because planes damaged in those areas did not make it home (Figure 1) (16). Employing similar logic, genomic regions that are not essential for cellular function or human health are more tolerable to genetic variation than those that are essential. In recent years, publicly available databases, such as the Genome Aggregation Database (gnomAD), have extensively cataloged the scope of genetic variation in the human genome and allowed us to gauge the potential ‘dispensability’ of genomic regions.
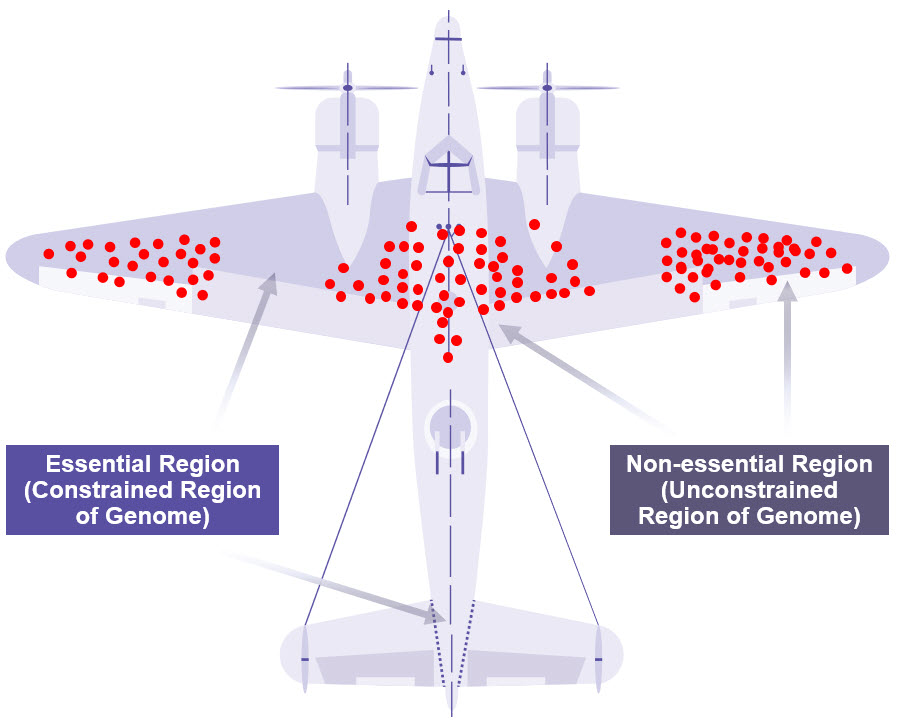
Figure 1: Warplane Model of Survivorship Bias
Genetic variants in the human genome can be looked at in the same way as Abraham Wald looked at bullet holes in warplanes (14). Observed mutations (red dots) often occur in unconstrained regions of the genome, which are less essential for survival and could be potentially ‘dispensable’. Credit: M. Grandjean (vector), McGeddon (picture), C. Moll (concept) (CC BY-SA 4.0).
Loss-of-Function Variants
Among the different types of genetic variants observed in the human genome, a small subset, known as LoF variants, lead to the inactivation of protein-coding genes. These variants provide important information about the phenotypic consequences of the loss of the gene. Traditionally, LoF variants have been viewed in the context of severe genetic diseases; however, LoF variants are not always deleterious. Historical medical case reports of incidental discovery of human ‘knockouts’ for specific alleles have been used to identify genes whose loss of function is tolerated in the human population. For example, benign LoF variation was first observed more than a century ago with the discovery of the variable blood group markers, the ABO antigens, that are known to determine blood type (17, 18). In this situation, a single-base deletion in the ABO gene encoding the glycosyltransferase protein results in a premature stop codon, lack of functional glycosyltransferase, and consequently creates the O blood type (17). We now understand the O blood type is not associated with any particular pathologic disease. Several studies have examined the association between blood groups and susceptibility to various diseases such as malaria (19), to suggest that in specific instances, human gene knockouts can be neutral or even provide an advantage in survival or health (20-22).

Figure 2: Schema for LoF Analysis
The essentiality of a gene can be gauged by evaluating its constraint metrics (pLI, LOEUF) in gnomAD. In unconstrained genes, heterozygous or homozygous LoF mutations can be observed, and their phenotypic consequences can be investigated using additional databases such as ClinVar (NCBI).
Identification and Validation of Dispensable Gene Editing Targets
Curated genetic databases such as gnomAD allow scientists to advance research efforts beyond medical case reports of gene ‘knockouts’ in humans to a more extensive screening of genes that harbor LoF variants (23-25). Genes with low evolutionary constraint are more likely to harbor LoF variants and can be identified using two primary metrics: the probability of loss-of-function intolerance (pLI) and the loss-of-function observed/expected upper bound fraction (LOEUF) score (9). Both metrics are derived from the ratio of the number of LoF variants observed when sequencing a gene to the number of variants predicted in that gene based on natural DNA mutation rates. Genes with low pLI (i.e. lower probability of a LoF resulting in haploinsufficiency, which is the occurrence of disease due to only one functioning copy of a gene) and high LOEUF score (i.e. higher tolerance towards LoF mutations) are more likely to be enriched for LoF mutations (9).
Within this subset of unconstrained genes, a powerful indicator of the dispensability of a gene is the presence of individuals that are LoF for both copies of the gene. An analysis of 19,197 genes in 141,456 individuals in gnomAD identified 1,815 genes that show biallelic inactivation (9). Although gnomAD is depleted for sequencing data from individuals with severe pediatric and genetic disorders, it is still possible that the identified LoF variants may have known disease associations. Therefore, variants that result in the homozygous deletion of a gene should be further cross-referenced in databases such as ClinVar and Genotype-Tissue Expression (GTEx) to screen for any disease-associated phenotypes. In Table 1 we provide data from gnomAD with examples of genes having varying degrees of constraint. From these examples, in cases of genes such as CD33, the presence of homozygous LoF individuals and lack of any known disease associations provides a strong genetic rationale for its dispensability. Conversely, constrained genes such as TP53 and CD47 do not have known LoF variants, are important for human health, and therefore unlikely to be suitable targets for genetic ablation. In the next section, we highlight how novel cell therapies can be developed by leveraging the genetically dispensable genes such as CD33.
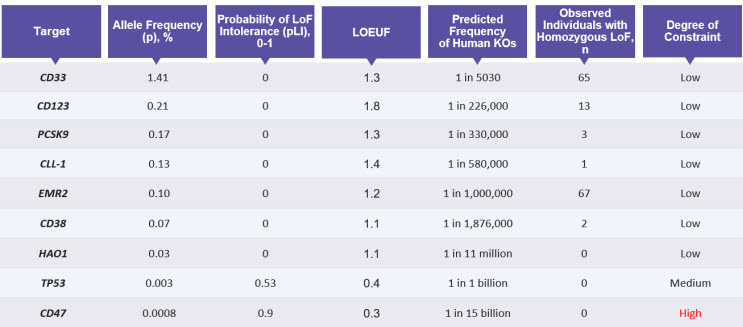
Table 1: Parameters to Evaluate the Degree of Constraint on Genes
Data from gnomAD helps predict which genes would be suitable targets for knockout, by analyzing the probability of intolerance to LoF mutations (pLI) and predicted frequency of LoF or knockouts (KOs) in humans. This table also provides the number of individuals likely to be homozygous for the loss-of-function for the listed genes.
Leveraging Human Genetics for Novel Cell Therapy Development: A Case Study
The absence of disease in an individual lacking functional copies of a gene gives confidence that removal of the gene from the human genome is well tolerated. This is the rationale behind Vor Biopharma’s novel cell therapy for treatment of AML, VOR33™. To date, treatment of AML with targeted therapies has been limited because the lack of tumor specific antigens on leukemia cells compared to normal cells results in “on-target, off-tumor” toxicity. An example of current AML treatment includes gemtuzumab ozogamicin (GO, Mylotarg™), which targets CD33 (26, 27), a member of the sialic acid-binding immunoglobin-like lectin (Siglec) family of cell surface proteins. While CD33 is highly expressed on AML cells, making it a promising immunotherapy target, its expression on healthy hematopoietic cells results in treatment related cytopenias owing to cytotoxic effects of therapies on non-malignant hematopoietic cells. Aiming to address limitations in current AML therapies, Vor Biopharma sought to engineer a therapeutic window to enable AML-specific therapies, while conferring protection to normal hematopoietic cells. To do this, we have engineered treatment-resistant hematopoietic stem and progenitor cells (HSPCs) through CRISPR/Cas9-mediated CD33 gene deletion from healthy, human leukocyte antigen-matched donor HSPCs for hematopoietic stem cell transplant into AML patients.
Genetic ablation of CD33 from HSPCs is supported by several lines of evidence that suggest dispensability of CD33 from the human genome. Kim et al. examined multiple databases of large population cohorts and identified individuals with naturally occurring homozygous LoF variants in CD33 (12). The existence of such individuals provides compelling evidence that CD33 is dispensable (9, 14-16). Additionally, an analysis of human genome sequencing data in gnomAD identifies 65 individuals who harbor homozygous LoF variation in CD33 (Table 1). Of these 65 individuals, the majority (85%) consists of a deletion of four base pairs in exon 3 (rs201074739), which results in a premature stop codon and complete loss of CD33 expression (28). The age distribution of homozygous carriers of CD33 LoF variants was found to be similar to that of heterozygotes and noncarriers, and at least four homozygous individuals were reported to have reached 64 to 93 years of age (29). Collectively, this evidence suggests that the therapeutic approach of removing CD33 from the human genome should have no detrimental effects on fitness and health. Since multiple receptors within the CD33-related Siglec family are expressed on hematopoietic cells (30, 31), the absence of phenotype upon CD33 LoF may be due to functional redundancy or compensation by other Siglec family members.
In addition to the genetic evidence, experimental studies demonstrate that human HSPCs and their progeny show no impairment of hematopoietic function when CD33 is removed from their genome (11, 12, 32). CD33 deficient human HSPCs demonstrated normal engraftment and differentiation in immunocompromised mice, while providing robust protection from the cytotoxic effects of CD33-directed companion therapeutics (11). Additionally, autologous CD33 knockout HSPC transplantation in non-human primates showed long-term multilineage engraftment of gene-edited cells that maintained normal hematopoietic function (12). In humans, the reconstituted hematopoietic compartment of patients receiving CD33-null HSPCs (VOR33™) is expected to be resistant to cytotoxicity induced by anti-CD33 directed therapies, such as GO. A first in human study testing this approach is currently enrolling patients (NCT04849910). In this clinical trial, the true biological dispensability of CD33 will be studied for the first time in a clinical setting examining endpoints related to engraftment. In the context of AML treatment, this strategy is anticipated to mitigate the hematological toxicity associated with GO and enable the testing of other pharmacologic and cellular therapies directed against CD33.
Importantly, the strategy of identifying dispensable gene editing targets through bioinformatic analyses of human genetic databases, and deleting them in normal hematopoietic cells to gain a therapeutic window, provides an important therapeutic framework for enabling tumor selective targeting by many of the cancer immunotherapies currently in development, thus improving patient quality of life and outcomes (Figure 3) (33).
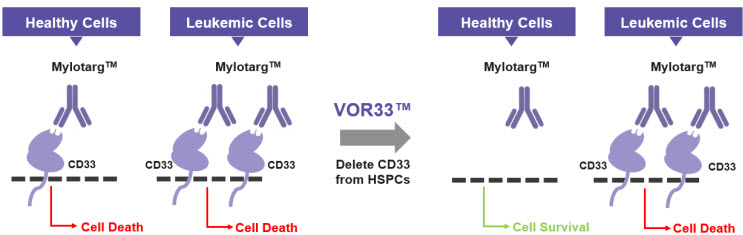
Figure 3: How VOR33™ Protects Healthy Cells
VOR33™ is developed by selectively deleting CD33 from healthy cells, which are then invisible to immunotherapies that target CD33 such as Mylotarg™. Healthy cells are spared, and leukemic cells are targeted, resulting in leukemic cell death. CD33, cluster of differentiation 33; HSPCs, hematopoietic stem progenitor cells.
Summary
Tackling the challenging and capital-intensive process of drug development requires novel ways of de-risking the therapeutic targets. The additional challenges and risks associated with genetic manipulation of cells for developing novel gene and cell therapies requires careful assessment of the underlying genetic targets. Leveraging human genetics data early in the drug discovery process to inform target prioritization is a powerful approach to mitigate risk and increase the likelihood of advancing safe therapies that make a transformative impact in patients’ lives. Here we outlined historical evidence of successful drug development based on understanding the naturally occurring genetic variation in humans. In particular, we focused on identifying and leveraging benign LoF variants for the development of novel cell therapies, such as VOR33™. These promising new therapies can alleviate the debilitating side-effects associated with many cancer immunotherapies and improve the quality of life for patients undergoing life-saving treatments. As genetic sequencing continues to expand, the identification of rarer LoF mutations will be an important factor in developing safer cell therapies for patients suffering from hematological malignances and other diseases.
References
- Dowden H, Munro J. Trends in clinical success rates and therapeutic focus. Nat Rev Drug Discov. 2019;18(7):495-6.
- Ochoa D, Karim M, Ghoussaini M, Hulcoop DG, McDonagh EM, Dunham I. Human genetics evidence supports two-thirds of the 2021 FDA-approved drugs. Nat Rev Drug Discov. 2022.
- Cook D, Brown D, Alexander R, March R, Morgan P, Satterthwaite G, et al. Lessons learned from the fate of AstraZeneca's drug pipeline: a five-dimensional framework. Nature Reviews Drug Discovery. 2014;13(6):419-31.
- Nelson MR, Tipney H, Painter JL, Shen J, Nicoletti P, Shen Y, et al. The support of human genetic evidence for approved drug indications. Nat Genet. 2015;47(8):856-60.
- MacArthur J, Bowler E, Cerezo M, Gil L, Hall P, Hastings E, et al. The new NHGRI-EBI Catalog of published genome-wide association studies (GWAS Catalog). Nucleic Acids Res. 2017;45(D1):D896-d901.
- Abifadel M, Varret M, Rabès JP, Allard D, Ouguerram K, Devillers M, et al. Mutations in PCSK9 cause autosomal dominant hypercholesterolemia. Nat Genet. 2003;34(2):154-6.
- Langsted A, Nordestgaard BG, Benn M, Tybjærg-Hansen A, Kamstrup PR. PCSK9 R46L Loss-of-Function Mutation Reduces Lipoprotein(a), LDL Cholesterol, and Risk of Aortic Valve Stenosis. The Journal of Clinical Endocrinology & Metabolism. 2016;101(9):3281-7.
- Karczewski KJ, Martin AR. Analytic and Translational Genetics. Annual Review of Biomedical Data Science. 2020;3(1):217-41.
- Karczewski KJ, Francioli LC, Tiao G, Cummings BB, Alfoldi J, Wang Q, et al. The mutational constraint spectrum quantified from variation in 141,456 humans. Nature. 2020;581(7809):434-43.
- Gene, Cell, & RNA Therapy Landscape: Q1 2022 Quarterly Data Report. asgt.org: American Society of Gene and Cell Therapy and Pharma Intelligence 2022.
- Borot F, Wang H, Ma Y, Jafarov T, Raza A, Ali AM, et al. Gene-edited stem cells enable CD33-directed immune therapy for myeloid malignancies. Proc Natl Acad Sci U S A. 2019;116(24):11978-87.
- Kim MY, Yu KR, Kenderian SS, Ruella M, Chen S, Shin TH, et al. Genetic Inactivation of CD33 in Hematopoietic Stem Cells to Enable CAR T Cell Immunotherapy for Acute Myeloid Leukemia. Cell. 2018;173(6):1439-53 e19.
- Godwin CD, Laszlo GS, Fiorenza S, Garling EE, Phi TD, Bates OM, et al. Targeting the membrane-proximal C2-set domain of CD33 for improved CD33-directed immunotherapy. Leukemia. 2021;35(9):2496-507.
- Havrilla JM, Pedersen BS, Layer RM, Quinlan AR. A map of constrained coding regions in the human genome. Nat Genet. 2019;51(1):88-95.
- Wallis WA. The Statistical Research Group, 1942–1945. Journal of the American Statistical Association. 1980;75(370):320-30.
- Mangel M, Samaniego FJ. Abraham Wald's Work on Aircraft Survivability. Journal of the American Statistical Association. 1984;79(386):259-67.
- Yamamoto F, Clausen H, White T, Marken J, Hakomori S. Molecular genetic basis of the histo-blood group ABO system. Nature. 1990;345(6272):229-33.
- Yamamoto F, Cid E, Yamamoto M, Saitou N, Bertranpetit J, Blancher A. An integrative evolution theory of histo-blood group ABO and related genes. Sci Rep. 2014;4:6601.
- Cserti CM, Dzik WH. The ABO blood group system and Plasmodium falciparum malaria. Blood. 2007;110(7):2250-8.
- Dahlén T, Clements M, Zhao J, Olsson ML, Edgren G. An agnostic study of associations between ABO and RhD blood group and phenome-wide disease risk. eLife. 2021;10:e65658.
- Anstee DJ. The relationship between blood groups and disease. Blood. 2010;115(23):4635-43.
- Ewald DR, Sumner SC. Blood type biochemistry and human disease. Wiley Interdiscip Rev Syst Biol Med. 2016;8(6):517-35.
- MacArthur DG, Tyler-Smith C. Loss-of-function variants in the genomes of healthy humans. Hum Mol Genet. 2010;19(R2):R125-30.
- Lim ET, Würtz P, Havulinna AS, Palta P, Tukiainen T, Rehnström K, et al. Distribution and Medical Impact of Loss-of-Function Variants in the Finnish Founder Population. PLOS Genetics. 2014;10(7):e1004494.
- Narasimhan VM, Hunt KA, Mason D, Baker CL, Karczewski KJ, Barnes MR, et al. Health and population effects of rare gene knockouts in adult humans with related parents. Science. 2016;352(6284):474-7.
- Norsworthy KJ, Ko CW, Lee JE, Liu J, John CS, Przepiorka D, et al. FDA Approval Summary: Mylotarg for Treatment of Patients with Relapsed or Refractory CD33-Positive Acute Myeloid Leukemia. Oncologist. 2018;23(9):1103-8.
- Appelbaum FR, Bernstein ID. Gemtuzumab ozogamicin for acute myeloid leukemia. Blood. 2017;130(22):2373-6.
- Papageorgiou I, Loken MR, Brodersen LE, Gbadamosi M, Uy GL, Meshinchi S, et al. CCGG deletion (rs201074739) in CD33 results in premature termination codon and complete loss of CD33 expression: another key variant with potential impact on response to CD33-directed agents. Leuk Lymphoma. 2019;60(9):2287-90.
- Sulem P, Helgason H, Oddson A, Stefansson H, Gudjonsson SA, Zink F, et al. Identification of a large set of rare complete human knockouts. Nat Genet. 2015;47(5):448-52.
- Crocker PR, Varki A. Siglecs, sialic acids and innate immunity. Trends Immunol. 2001;22(6):337-42.
- Brinkman-Van der Linden EC, Angata T, Reynolds SA, Powell LD, Hedrick SM, Varki A. CD33/Siglec-3 binding specificity, expression pattern, and consequences of gene deletion in mice. Mol Cell Biol. 2003;23(12):4199-206.
- Humbert O, Laszlo GS, Sichel S, Ironside C, Haworth KG, Bates OM, et al. Engineering resistance to CD33-targeted immunotherapy in normal hematopoiesis by CRISPR/Cas9-deletion of CD33 exon 2. Leukemia. 2019;33(3):762-808.
- Gill S, Tasian SK, Ruella M, Shestova O, Li Y, Porter DL, et al. Preclinical targeting of human acute myeloid leukemia and myeloablation using chimeric antigen receptor-modified T cells. Blood. 2014;123(15):2343-54.