
How To Overcome Potency Assay Development Challenges For Gene Therapies
By BioPhorum
 Gene therapies (GTs) are a complex class of biopharmaceuticals that offer the prospect of curative treatments for diseases and genetic disorders. Critical quality attributes (CQAs) such as potency should be identified and controlled to ensure the desired product quality and, thus, safety and efficacy.
Gene therapies (GTs) are a complex class of biopharmaceuticals that offer the prospect of curative treatments for diseases and genetic disorders. Critical quality attributes (CQAs) such as potency should be identified and controlled to ensure the desired product quality and, thus, safety and efficacy.
Potency is one of the CQAs for GT development and manufacturing that is most related to the therapeutic product’s mechanism of action (MoA). However, developing and validating appropriate potency assays that reflect the MoA acceptable to regulators is a process fraught with challenges.
What Is Meant By The Term Potency?
The FDA has defined potency as “the specific ability or capacity of the product, as indicated by appropriate laboratory tests or by adequately controlled clinical data obtained through the administration of the product in the manner intended, to effect a given result.”
Tests to indicate potency consist of either in vitro or in vivo tests (or both) that have been specifically designed for each product. Potency is a key measure of the ability to establish manufacturing consistency, demonstrate the comparability of a product following manufacturing changes, evaluate product release and stability, and ascertain shelf life.
Gene Therapy Modalities
Whereas cell therapy involves the transfer of cells with the relevant function into the patient, GTs involve the transfer of genetic material and the uptake of the gene into the appropriate cells of the body. GT products can be subcategorized based on the nature and route of delivery (in vivo versus ex vivo) of the delivery vehicle.
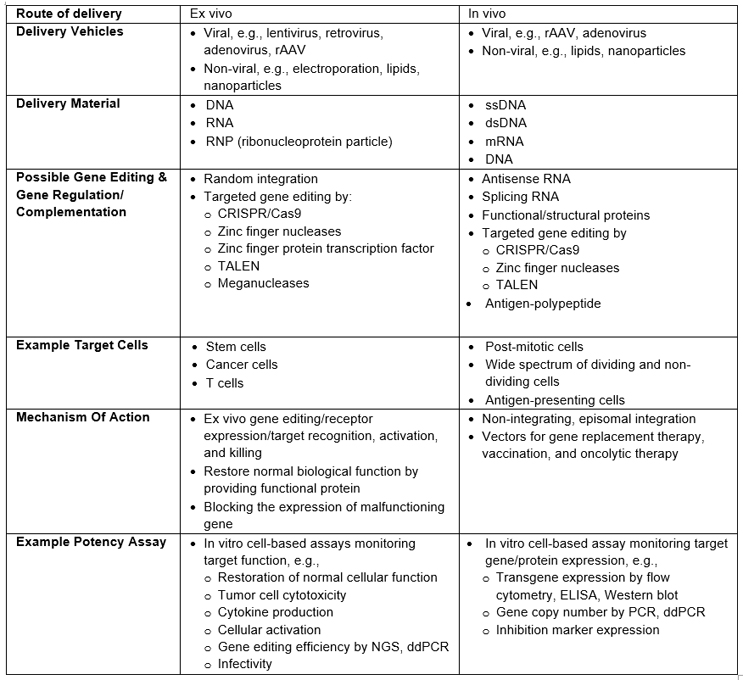
The approach to potency assays discussed below can be applied to these GT modalities. Table 1 provides a high-level review of common in vivo and ex vivo GT processes, with examples of commonly used assays to measure potency. Note: It does not represent a comprehensive review of all GT modalities or clinical or commercial readiness.
Table 1: Examples Of Processes, Methodologies, And Potency Assays For In Vivo And Ex Vivo GTs
Challenges For Gene Therapy Potency Assay Development
For traditional biopharmaceuticals (e.g., protein biologics), the potency of the bioactive agent is often demonstrated by a single, quantitative biological assay reflecting the MoA. However, for GT developers, there are some unique complexities associated with these products that increase the challenge to develop potency assays.
For example, GT products require multiple coordinated steps. In the case of viral therapeutics, the MoA includes:
- The delivery of the genetic information (payload) for the bioactive molecule to the target cells.
- The production of the bioactive molecule by the target cells.
- The biological effect of the bioactive molecule.
The Matrix Approach
Considering the additional complexities of GT products, it may be more appropriate to consider an incremental approach to developing and implementing potency tests, i.e., a “matrix approach.” In March 2022, the FDA advocated the use of a matrix approach to measure potency.
In the discovery and preclinical phases, sophisticated transgenic animal models are typically used to show a proof of concept that mirrors the MoA. However, in vivo potency assays commonly lack robustness, reproducibility, and the precision required for GMP assay validation, making them undesirable in the long term as part of the control strategy. Consequently, the introduction and continuous optimization of an in vitro potency testing strategy, covering correct intracellular processing of transgenic payload in the cell matrix and the functionality of the transgenic products, is recommended for GTs, starting in the early phases of clinical development.
Potency assays need not capture every MoA, only the key ones. The matrix approach involves capturing two or more MoAs and compiling the data to draw a collective conclusion. This approach combines complementary assays based on product-specific attributes covering biological or non-biological methods. Potency methods should be MoA-reflective, though it is important to be aware that potency may not be able to be correlated with in vivo efficacy and clinical outcome.
The sum of the multiple assays (matrix of assays) should demonstrate the intracellular processing of the recombinant genetic information in the target cells and, in the best case, up to its functionality in the target cells. This intracellular processing can be shown experimentally in vitro or in vivo. At least one assay of this matrix used to measure potency should be quantitative and expressed in units of activity calibrated against an appropriately qualified reference standard. Where no qualified reference standard exists, a well-characterized assay control is recommended to monitor assay performance only.
Regulators (specifically, the FDA and the EMA) recommend establishing multiple potential potency assays as early as possible during initial clinical phases to eventually identify and qualify the best potency test(s) for pivotal trials; this is the beginning of the matrix approach.
During preclinical and early clinical development, the selection of potency assay methods is guided by the presumed MoA and should ensure the correct functional activity (see Table 1 for examples). For GTs, we recommend following a phased approach, appropriate to assay validation per International Council for Harmonisation guidelines.
In general, early-phase specifications are established to ensure that materials used in early clinical studies are of appropriate product quality to reduce the risk of harm to patients. One of the key challenges in setting specifications in early development for GTs is the limited data available when only one or a few lots have been manufactured. Also, due to the complexity of GT drug products, there are often multiple MoAs and the timeframe for investigating MoAs is often shorter for GTs than for protein biologics. The availability of data generated by the product characterization at the preclinical phase and the level of manufacturing experience at early stages may be even less in some instances in the GT field.
Consequently, specification setting for GTs is more reliant on molecule-specific information acquired during development phases. However, the general principles of the phase-dependent specification and criteria settings for GTs are conceptually similar to those for protein biologics.
Multiple assays should be developed to measure potency (see Table 1 for example), but not all will be deemed suitable for submission as evidence for licensure – orthogonal methods may support characterization of cell function during product development or support comparability studies, for example, but may not be suitable candidates for commercial lot release. Therefore, it is to be expected that some assays may be set aside during preclinical and early clinical phase studies. The final matrix of assays selected for commercial lot release need to be fully validated before applying for licensure. A correlation assessment should be performed for submissions that shows the correlation of potency assay results to relevant product-specific biological activity and refers to genetic data from preclinical or clinical work. The analytical assays from preclinical activities can be used informally as (extended) characterization methods to provide better product understanding for monitoring purposes, comparability assessment, or the briefing package. The assay should be assessed for accuracy, precision, sensitivity, robustness, system suitability, and specificity.
Mimicking the biological activity in the clinical situation is not always necessary; a correlation between the expected clinical response and the activity in the biological assay can be established in pharmacodynamic or clinical studies.
The cell line selected for the in vitro potency assay should be adequate to reflect the MoA, which may be more challenging for ex vivo GTs than for in vivo GTs.
Potency Assays And Readouts For In Vivo Gene Therapy
The ideal potency assay should reflect the key MoA while providing quantitative data to support lot conformance, stability, manufacturing consistency, and comparability. For in vivo GT products, in vitro potency assays that reflect MoA would be based on transduction of a selected cell type representative of the target organ or tissue. This is followed by measuring the functional effect or activity of the expressed transgene sequence in a dose-dependent manner, preferably with the potency reported in units relative to a reference standard.
Because transgene sequences are different for every product, measuring the functional effect can vary greatly and depends on the exact nature of the transgene. In cases where the transgene sequence may have multiple effects (e.g., scaffolding proteins), potency assays may be designed to measure one or more of its effects.
Potency Assays And Readouts For Ex Vivo Gene Therapy
For ex vivo gene-modified cell therapy products (referred to as ex vivo GT), potency is an essential aspect of the quality control system for both drug substance (where applicable) and drug product. Potency assays are performed to help assure the strength and stability of products used throughout the clinical studies process, as well as for licensed products.
There are many challenges during the manufacturing development process of these more patient-specific therapies. The drug product for this type of therapeutic (i.e., engineered CAR T cells) can be sourced from a healthy donor (allogeneic) or from and for individual patients (autologous). Determining CQAs, understanding sources of variation, identifying critical steps in the manufacturing process, and establishing an integrated control strategy are all essential to ensure consistent product quality before product release and administration to patients.
Conclusion
GTs have the potential to provide significant patient benefits and revolutionize curative treatments. Manufacturing these products involves various processes that require intensive and sophisticated testing where potency is a key test included in the control strategy. Developing potency assays for GTs is highly complex and is compounded by additional challenges uniquely and commonly encountered by GT developers.
The GT field is evolving rapidly with new technologies and scientific discoveries. We expect that potency assays would need to evolve accordingly as knowledge and understanding of GT products are accumulated via ongoing development and clinical studies and with the availability of appropriate technologies suitable for the purpose. There is a growing need for a greater understanding of structure-function relationships of GTs to develop potency assays further. Emerging analytics, such as next-generation sequencing, could be transformed into a quality control-friendly assay for GTs. Alternative methods that may not yet exist will emerge to contribute to a better understanding of potency in the future.
This article is a summary of a recent BioPhorum publication on the topic. To read more, check out the full report, Challenges for potency assay development for in vivo and ex vivo gene therapies and the matrix approach.