
FDA User Fee Programs Reauthorized: FDA's CBER Is A Clear Winner
By Nancy Bradish Myers, Catalyst Healthcare Consulting, Inc.

After years of planning, negotiation, and input, the user fee reauthorization bill was passed and signed into law on the very date that the programs were scheduled to sunset. To many FDA watchers’ surprise, politics dictated that the bill moved free of the significant number of policy riders that Members and some interest groups were considering as likely attachments (riders) to the must-pass reauthorization effort, which in addition to PDUFA includes GDUFA (generics), BsUFA (biosimilars), and MDUFA (devices).
With this being the most squeaky-clean reauthorization seen in 30 years, the news with its passage is in the agreement letters, rather than what was attached to them. The big winner of PDUFA VII is the Center for Biologics Evaluation & Research (CBER), and events of the last few days indicate CBER is ready with a plan.
CBER is poised to grow dramatically. In the first year of PDUFA VII, the target of 132 new hires is a quadrupling of the 32 FTEs funded under all of PDUFA VI. And the five-year total of 228 new FTEs under PDUFA VII is over seven times the total targeted hires for all of PDUFA VI.
With these new employees will come significant new user fee funding to advance innovative pilot projects, develop new guidance and increase outreach and interactions with industry. As I noted in an article for Pharmaceutical Online last year, CBER’s budget will increase by $348.7 million over five years.
Staffing: Quadruple the Hiring Goal in First Year of PDUFA VII (132), Vs. All of PDUFA VI (32)
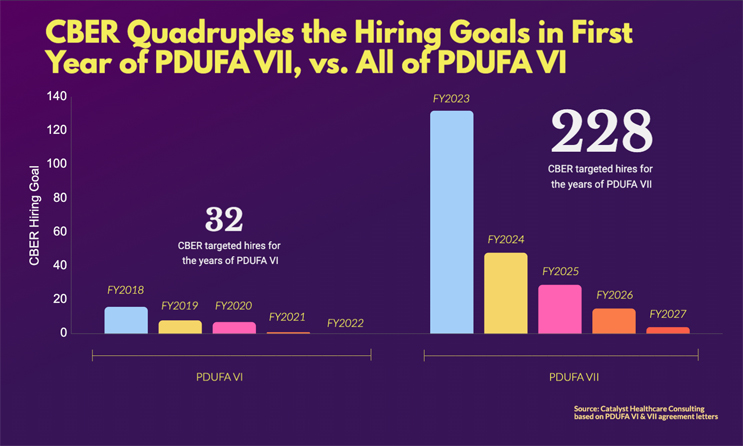
Source: Catalyst Healthcare Consulting, PDUFA VI & VII agreement letters
CBER’s Reorganization Plan Shows An Effort To Prepare For The Future
Demonstrating CBER’s readiness to seize the opportunities created by PDUFA VII, just two days after the House and Senate reached agreement on a bill, CBER formally offered more details about this significant reorganization of its gene & cell therapy review office in the Federal Register. This reorganization has been blessed by the administration’s Office of Personnel Management and Congress.
CBER Creates A New Super Office: Office Of Therapeutic Products (OTP)
Over the past few years, FDA has created “super offices” within its centers to facilitate cross-functional work within and across centers. A few examples of existing super offices are the Oncology Center of Excellence, CDER’s Office of Pharmaceutical Quality, and CDRH’s Office of Digital Health and Office of Product Evaluation and Quality. Two common threads among these super offices are a vision to build reasonable cross-office management controls and to prepare to lean into significant growth in an area of anticipated innovation.
As a practical matter, OTP, which will soon total 455 to 460 employees, has been created to help manage this growing part of CBER, create more product-specific offices, and develop a matrixed organization to drive efficiency and facilitate innovation. Lowering the number of employees per manager is also anticipated.
But more importantly, this OTP super office demonstrates a leadership dedication to move cell and gene therapies into the mainstream. This new structure, coupled with the user fee agreement language, will necessitate development of an innovative policy shop that will explore ways to lean into accelerated approvals for gene therapies and adoption of experimental efforts, as well as create pilots that allow for increased sponsor/agency communication opportunities successfully tested during Operation Warp Speed.
FDA has retained an outside recruiting firm to conduct a national search to fill the OTP director position, which will likely serve as an unofficial deputy center director role. The OTP director will report directly to CBER Director Peter Marks.
It is said the vision of this effort has been built to leverage the successes of the Oncology Center of Excellence run by Dr. Rick Pazdur, which is staffed by a nucleus of tremendously devoted and talented staff working to advance cancer care for patients. Dr. Marks believes this super office will attract significant additional talent.
OTP will house new and reorganized offices, run by an increasing number of office directors and managers to ensure review consistency and to focus on team building. Although current staff can apply for these roles, it is anticipated that external experts with a diversity of experience, including industry bona fides, will be sought to build on current expertise.
New, more defined product-specific offices include:
- Office of Gene Therapy CMC
- Office of Cell Therapy and Human Tissues CMC
- Office of Plasma Therapy
Cross-functional offices with potential SWAT Team staffing potential include:
- Office of Clinical Evaluation
- Office of Pharmacology/Toxicology
- Office of Review Management and Regulatory Review
The reorganization is intended to create “flexibility and capacity for future growth” in FTEs and workload, “avoiding the need for continual reorganizations,” CBER has said. The changes will “position OTP to focus on commitments,” including those in the user fee agreement. For example, the larger, re-envisioned Office of Clinical Evaluation will allow teams of reviewers to move more fluidly and be assigned to areas of increased need and priority.
It is noteworthy that adding an Office of Cell Therapy and Human Tissue CMC demonstrates CBER’s recognition that this is an area of anticipated growth, in which there is a distinct need for expertise.
Overall, the super office will “improve functional alignment, increase review capabilities and enhance expertise on new cell and gene therapies,” CBER has said. The reorganization, announced Sept. 28, went into effect on Sept. 16.
Given these changes, it’s clear that CBER may be preparing the new OTP to optimize the pace of product reviews in the cell & gene therapy space.
With Much To Be Done, CBER Will Pace Itself
Before the cell and gene therapy industry gets too excited, we should note the need for patience.
Strategic hiring at FDA takes time and attracting the right candidates to work for the federal government can be difficult, even with 21 Century Cures Act hiring authorities and increased pay scales. It is noteworthy that CBER could not fill all the positions it was granted in the last round of user fee negotiations, PDUFA VI.
Then, once FDA hires new staff, training takes even more time. CBER leadership has said it requires about two years to fully train a new CBER reviewer. So, while the new hiring authority provides a much-needed boost, it will take some time for the full effect to be felt and reflected in interactions with innovators.
All this will be compounded by the slow return of FDA staffers to White Oak post-pandemic and the need for management flexibility and novel approaches to mentoring. This will require oversight.
In addition to hiring, CBER has agreed to launch a significant number of new initiatives in return for increased user fees. A few agreed-upon actions of interest focused on facilitating product development and review for gene & cell therapies are:
- Hold a patient-focused drug development (PFDD) meeting on patient experience and preference data on cell & gene therapy products – by end of FY 2023.
- Issue draft guidance on evaluation of efficacy in small patient populations using novel trial designs and statistical methods.
- Issue Q&A draft guidance on common issues faced by sponsors - by end of FY 2024.
- Hold a public meeting to solicit input on methods and approaches for capturing post-approval safety and efficacy data to inform draft guidance - by end of FY 2025.
- Update the guidance on Expedited Programs for Regenerative Medicine Therapies for Serious Conditions (including CMC considerations) - by the end of FY 2025.
- Accelerate CBER IT modernization: Establish a multiyear modernization road map, including concrete implementation phases and milestones - by the end of Q4 FY 2022.
A new Type D meeting created in the user fee negotiations will also likely be a welcome addition for gene & cell therapy developers. Under the agreement, sponsors may request this new meeting to focus on no more than two issues that do not require input from more than three disciplines/divisions. These meetings are intended to address narrow questions on which a sponsor is seeking guidance, a follow-up question that raises a new issue after a formal meeting, or a general question about an innovative development approach that does not require extensive, detailed advice.
In addition, a formal Request for Clarification process will now be available to sponsors. This is intended to improve follow-up communication after receiving written responses or meeting minutes from FDA.
CBER will also act in chemistry, manufacturing, and controls (CMC), which is a key challenge in development of cell & gene therapies. For example, to promote reviewer use of Four-Part Harmony in CMC information requests, CBER will update its SOPP (standard operating procedures & policies) on the topic by the end of FY 2023.
Completely Clean Bill Today Means Potential Legislative Changes On The Horizon
Though we will hail this round as a clean reauthorization, Congress must revisit several important programs only temporarily extended with the continuing resolution. While the programs typically move with the UFAs, the following programs will sunset in mid-December:
- Best Pharmaceuticals for Children Act, which encourages pediatric studies by granting an additional six months of patent exclusivity
- Humanitarian Device Exemption, which exempts developers from efficacy testing requirements for devices intended to treat rare diseases
- Pediatric device consortia program – provides resources, which can include funding, to help guide and encourage development of medical devices for pediatric patients
- Market exclusivity for drugs containing single enantiomers (Hatch-Waxman Improvement Act of 2022)
- Critical Path public-private partnership, a longstanding initiative to modernize drug development and manufacturing and facilitate translation of scientific advances into commercial products
- Reporting requirements related to pending generic drug applications and the number subject to priority review, with details such as the number awaiting action by the applicant, or by the agency, and the number approved by FDA
- Orphan drug grants program, under which FDA funds investigators developing products for rare diseases
Delayed Policy Considerations
Separate from these legislative reauthorizations, following the November mid-term elections, the outgoing Congress may use the opportunity for a second bite at the “adding more policy changes” apple. Some of these changes would affect gene & cell therapies. Those with enough support to potentially be attached to legislation include:
- Accelerated approval reform (requiring post-approval studies to be underway at approval, more frequent reporting, streamlined withdrawal procedures)
- Clarifying that orphan drug exclusivity applies only to the specific indication or use (e.g., adult or pediatric)
- Provisions in the VALID Act, which would create a risk-based, tiered system of FDA oversight of laboratory-developed tests (LDTs)
- Efforts to limit animal testing in drug development – this would end animal testing mandates, allowing sponsors to engage in alternatives where appropriate
Conclusions
At the final hour, the PDUFA VII user fee reauthorization is complete and will bring significant funds and growth to specific programs within FDA to supplement Congressional appropriations. Now it’s “go time” for the beneficiaries, in particular, CBER, to quickly start implementing the array of new commitments. I expect we will hear more about the CBER reorganization over the next few weeks. And as we head into December, get ready for continued debate on policies and programs important to FDA. We are likely to see Congress revisit several legislative rider initiatives that would affect the agency overall.
About The Author:
 Nancy Bradish Myers, JD, founder and CEO of Catalyst Healthcare Consulting, Inc., is a Washington, D.C.-based attorney with deep expertise in healthcare law and regulation, policy development, patient engagement, and government relations. She served as senior policy counsel in the Office of the FDA Commissioner and as special assistant/senior strategic advisor to the FDA acting deputy commissioner for operations. Myers has been closely involved in drug, biotechnology, and medtech regulatory issues for over 25 years. She advises cutting-edge clients on regulatory and strategic policy matters. You can reach her at nancy@catalysthcc.com or connect with her on LinkedIn.
Nancy Bradish Myers, JD, founder and CEO of Catalyst Healthcare Consulting, Inc., is a Washington, D.C.-based attorney with deep expertise in healthcare law and regulation, policy development, patient engagement, and government relations. She served as senior policy counsel in the Office of the FDA Commissioner and as special assistant/senior strategic advisor to the FDA acting deputy commissioner for operations. Myers has been closely involved in drug, biotechnology, and medtech regulatory issues for over 25 years. She advises cutting-edge clients on regulatory and strategic policy matters. You can reach her at nancy@catalysthcc.com or connect with her on LinkedIn.