
Best Practices In Preclinical Development Of Cell Therapy Products
By Nathan Manley and Katy Spink, Dark Horse Consulting Group, Inc.

The cell therapy field is currently in an exciting era of expansion, with a large number of candidate therapies advancing into clinical testing and many more progressing through preclinical pipelines. As the number of early-stage candidate cell therapy products continues to increase, so does the need to define effective preclinical development strategies that support clinical translation. Given the diverse and often multifunctional nature of cell-based therapeutics, it is not possible to define a one-size-fits-all preclinical program for cell therapy. Instead, preclinical studies must be customized to support the feasibility of the proposed administration route and appropriate application procedure, address the candidate product’s specific therapeutic properties, and fully characterize any potential safety concerns.
For a cell therapy preclinical program to be successful, several unique challenges must be addressed. First, while questioning the clinical relevance of existing preclinical models is nothing new, this concern carries added weight when considering that many cell therapy products are likely to have multiple mechanisms of action, each of which may be more or less faithfully reproduced in any given model system. Second, because many cell therapies are designed for long-term or even permanent host engraftment, preclinical studies often need to support a long in-life phase to appropriately represent the product’s potential therapeutic benefit and uncover potential safety concerns. In addition, cell therapies that intend to use a novel clinical delivery device may need to include feasibility and safety testing in a large animal model. Finally, a critical consideration for preclinical testing of human cell therapy products is whether a host immune response poses a safety concern or, in the case of immunotherapy products, is adequate. While no single set of solutions exists to address these challenges, ongoing progress in the field has uncovered general strategies to streamline preclinical efforts and enhance their translatability.
Drawing from current regulatory guidance documents and industry thought leaders, this article provides an overview of the preclinical development process for cell therapy products, discusses effective strategies to improve program efficiency, and outlines key regulatory requirements that must be addressed by preclinical studies. Given their criticality to program success, special attention is given to the topics of preclinical model selection, the design and execution of pivotal preclinical proof of concept (PoC) efficacy and safety studies, and the optimal timing for regulatory engagement during preclinical development.
From Bedside To Bench And Back Again
A common misconception of cell therapy preclinical development is that it is part of a linear journey from bench to bedside. In reality, it is often the case that after preclinical development has initiated, the product’s intended clinical use inevitably evolves. Instead, use of a circular approach — one that begins with identification of the product’s intended clinical application, followed by a developmental program that is guided by and ultimately arrives back at the intended clinical use — helps avoid wasted preclinical efforts and streamlines overall cell therapy product development.1 An effective way to implement this bedside to bench and back again approach is to draft a target product profile (TPP) as early as possible.
The TPP is instrumental in guiding preclinical efforts because it defines key elements of a cell therapy product’s intended clinical use, including patient inclusion/exclusion criteria, the method and timing of treatment administration, dose, potential safety concerns, and the product’s expected mechanism of action (MoA).2 Establishing a draft TPP prior to preclinical development aids selection of an appropriate preclinical model system for drug product efficacy and safety testing, guides preclinical study design, and identifies knowledge gaps, such as optimal therapeutic dose or MoA, that should be addressed during preclinical development. Although investigators often are reluctant to draft a TPP when faced with many unknowns early in development, an alternative approach is to identify the cell therapy product’s potential attributes, ranging from the ideal scenario to the minimum acceptable criteria needed for the product to be viable. Ultimately, the TPP guides the creation of the drug product’s clinical use label; early efforts to draft a TPP can help ensure that subsequent preclinical and product development efforts are conducted in a manner that are maximally aligned with the product’s purpose.
Efforts to define the intended purpose and target market for a candidate cell therapy product should also explore current standard of care (SoC) treatments. Because some of these existing treatments will be the competition, their relative cost, ease of administration, and impact and durability on patient outcome are key comparators that will influence market adoption of the new cell therapy product.3 Cell therapies that are expected to have a high cost and/or require a complicated administration procedure will have a higher bar for efficacy; to compete with cheaper/easier SoC options, the candidate cell therapy must offer a substantially greater therapeutic benefit to patients. These trade-offs should be carefully considered while crafting the product’s TPP. As discussed further below, preclinical studies can provide an early read on how a new candidate therapy will compare to or, in some cases, even synergize with current SoC treatments. By carefully mapping the intended clinical path for a new cell therapy product, its preclinical program can be appropriately tailored to address key questions and support successful adoption into the clinical market.
Choosing The Right Preclinical Model System(s)
Given the highly specialized and complex functional nature of cell therapy products, the selection of appropriate model systems for efficacy and safety testing is crucial for effective preclinical development. There is no regulatory precedent for selecting among the different types of preclinical model systems; instead, the goal of the investigator is to identify the model system(s) that most accurately recapitulate(s) critical elements of their cell therapy product’s intended clinical use (e.g., as defined by the TPP) and to confirm the suitability of their choice with the appropriate regulatory authority.4-5 With the rapid expansion of publicly available preclinical and clinical cell therapy data, the potential for new cell therapy programs to cross-reference existing data should be explored as a first step in preclinical planning and may point to a smaller subset of required de novo studies. In addition, in vitro systems may be more appropriate than in vivo animal models for generating certain kinds of preclinical efficacy or safety data if, for example, product efficacy requires interaction with human ligands/cells (e.g., chimeric antigen receptor-based and related immunotherapies) or if product safety is based on the absence of a residual cell population whose detection requires in vitro expansion (e.g., residual proliferative cells in an irradiated cell therapy product). Replacement of animal testing with preexisting data or in vitro studies should be explored whenever possible, both as a means to increase program efficiency and to lessen the burden on animal life.6
Typically, cell therapy products require some degree of in vivo testing to fully characterize preclinical efficacy or safety. Included among the key drivers of in vivo model selection are 1.) embodiment of relevant anatomical, physiological, and biochemical parameters of the target indication, 2.) administration parameters, such as cell dose, dose frequency, and the timing/method of delivery, and 3.) the ability to measure clinically relevant efficacy and safety endpoints. In terms of how best to address these considerations, an important question is whether small or large model systems (or a combination of the two) are needed. Small animal models (e.g., mice and rats) offer several advantages, including a wide range of transgenic disease/injury models, the ability for large study cohort sizes due to relatively low cost/husbandry needs, and, importantly, the availability of immunodeficient strains that can support long-term engraftment of human cell therapy products. Large animal models (e.g., pigs, sheep, rabbits, dogs, and non-human primates), while more limiting in the above attributes, instead offer the ability to test clinically relevant cell doses using the intended clinical administration device and in many cases, provide a closer representation of humans with respect to organ anatomy and injury/disease pathophysiology; this is important because large animal models can potentially provide greater predictability of clinical safety and efficacy for cell therapy products.7-9 Beyond the question of small versus large, another consideration is whether homologous modeling — testing the animal-derived equivalent of a human cell therapy product — is required. Homologous modeling may be needed for human cell therapy products whose postulated MoA depends on the presence of a human-specific factor (e.g., many immunotherapies) or for which long-term engraftment is anticipated and required in humans but cannot be sufficiently demonstrated in animals.
Given the product-specific nature of preclinical model selection, it is highly recommended that investigators seek regulatory feedback once they have identified the preclinical model(s) considered best-suited to addressing the unique efficacy and safety attributes of their candidate cell therapy product. Obtaining regulatory buy-in early in the development process (e.g., through an INTERACT meeting with the U.S. FDA10) can help identify pilot studies needed to confirm suitability of the chosen preclinical model(s) and ensures that the product’s overall preclinical strategy is well aligned with regulatory expectations.
Once an appropriate preclinical model system has been identified, investigators should then conduct pilot studies to tailor the model to the specific testing needs of their cell therapy product. For example, pilot testing may include calibration of injury/disease severity, such that detection of product efficacy is properly balanced with the long-term survival and well-being of animal subjects. Pilot testing may also be used to conduct preliminary dose escalation studies and refine the timing, method, and frequency of product administration. For cell therapy products that are expected to exhibit long-term clinical persistence, pilot testing also can allow optimization of any additional immunosuppressant treatments that may be needed to support durable engraftment. Finally, pilot studies can be used to pressure test candidate efficacy and safety endpoints and provide supportive data to statistically power subsequent PoC efficacy and pivotal safety studies. With the right preclinical model in place and additional information gathered from pilot testing, PoC efficacy and safety studies can then be designed to provide maximal support for a cell therapy product’s subsequent advancement to clinical testing.
Designing Pivotal Preclinical Studies
The final section of this article provides an overview of general guidelines and key requirements for designing pivotal preclinical PoC efficacy and safety studies for cell therapy products. Because patient safety is the primary objective of initial clinical testing, pivotal preclinical safety studies generally must adhere to stricter guidelines than PoC efficacy studies with respect to design and execution. Conversely, preclinical PoC study design should generate efficacy data that is clinically meaningful and provides insight into the cell therapy product’s therapeutic MoA. Given these distinct objectives, there are several key factors that must be considered when designing each type of pivotal preclinical study.
First, careful attention must be paid to preclinical design logistics, such as study endpoints, cohort sizes, control groups, and in-life duration, since these parameters impact the interpretability and clinical relevance of the resulting efficacy and safety data. For PoC efficacy studies, cohort sizes must be statistically powered to detect treatment-associated changes in one or more predetermined outcome measures relative to an appropriate control group. At a minimum, control treatment should consist of test vehicle (TV) administered in the same manner as the test article (TA). Clinical relevance may be enhanced by including SoC treatment applied to a separate cohort for comparison (and potentially in combination with the TA, if clinically appropriate). The in-life duration of PoC efficacy studies should be sufficiently long to demonstrate sustained benefit that is likely to translate to improved patient outcome and also should include sampling that will allow further characterization of the cell therapy product’s hypothesized MoA(s). In the case of pivotal safety studies, endpoints should characterize the short- and long-term in vivo biodistribution of the cell product using a clinically relevant delivery method and assess systemic and local toxicities associated with the treatment or administration procedure.5 As such, cohort size for safety testing is determined by the expected frequency of occurrence for any potential safety concerns, typically requires testing of multiple TA lots in both male and female appropriately aged subjects, and includes a control group administered TV. The in-life duration and sampling of pivotal safety studies is determined by the expected in vivo persistence and proliferative status of the administered cells and must be sufficiently long to assess the potential for relevant long-term safety concerns, such as tumor or ectopic tissue formation. In the case of pluripotent stem cell-derived products, separate tumorigenicity studies involving PSC-spiked TA may also be required.
The criteria for TA lot selection and dosing also can be different for pivotal PoC efficacy and safety studies. PoC efficacy studies should use TA lots that are comparable to the intended clinical cell therapy product, both with respect to its manufacturing process and product attributes (with any relevant differences noted). In contrast, TA lots used for pivotal preclinical safety studies must be generated using the clinical manufacturing process (though it need not be in accordance with current good manufacturing practices) and must meet the intended product specifications that will be used for initial clinical release. PoC efficacy study design should incorporate dose response testing (i.e., the proportional target clinical dose plus one or more non-efficacious doses), whereas safety studies should include a dose that is proportionally equivalent, or in excess of, the maximum intended clinical dose to sufficiently address product toxicity. Importantly, given the likelihood that product characterization methods will evolve and improve during product development, sufficient retains of each TA lot used in pivotal preclinical studies should be maintained for use in future analytical development and comparability testing (ideally, TA used in pilot studies as well).
Finally, there are differences in the degree of quality oversight and documentation required for pivotal preclinical efficacy and safety studies. Safety studies, but not PoC efficacy studies, typically must be conducted in accordance with current good laboratory practices and with the corresponding quality oversight.11 In addition, all results generated by pivotal safety studies must be documented in an audited study report containing a complete account of the study data in tabular format.
As discussed above, the relative timing of obtaining guidance from the relevant regulatory agency is a critical component of a successful cell therapy preclinical program, and this is particularly true for later stage, pivotal studies. The data generated by pivotal preclinical studies plays a crucial role in the risk-benefit analysis that will determine whether a candidate cell therapy is ready for clinical testing. Given this, and the significant impact on program costs, timelines, and animal life, it is imperative that investigators obtain regulatory buy-in on the design and endpoints of all clinical-enabling preclinical safety studies (e.g., through a pre-IND meeting with the FDA) prior to their execution. Ideally, the timing of this regulatory interaction should follow lockdown of the Phase 1 clinical manufacturing process, identification of the analytical testing methods that will be used to characterize and release clinical grade product, and generation of protocol synopses for the key studies that will be used to assess preclinical efficacy and safety of the candidate cell therapy product.
Conclusions
Taken together, the principles described here provide an overview of best practices in preclinical development for cell therapy products. A successful preclinical program starts with, and is guided by, an understanding of the candidate product’s intended clinical use and is further strengthened by regulatory engagement at key stages of development (Figure 1). Similarly, preclinical model selection and study design should be optimized to address the specific efficacy and safety attributes of the candidate cell therapy product. As the number of innovative cell therapy products continues to increase, implementation of a robust and clinically guided preclinical program can help to ensure that more patients are able to benefit from the transformative potential of this exciting field.
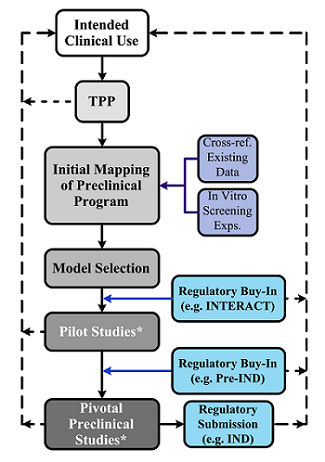
Figure 1: Preclinical Development Path of Cell Therapy Products – The optimal preclinical development path for a cell therapy product begins by defining its intended clinical use and generating a draft TPP. Subsequently, preclinical development progresses through initial program (guided by existing clinical or preclinical data and preliminary in vitro screening experiments), model selection, pilot studies, and lastly, clinical-enabling, pivotal studies. Blue boxes denote key stages of regulatory engagement, including regulatory buy-in following preclinical model(s) selection and prior to the execution of pivotal studies. The cell therapy product’s intended clinical use is further shaped and refined throughout the preclinical development process; key points of feedback from preclinical activities to the intended clinical use are denoted by dotted arrows. Asterisks denote preclinical activities that should generate TA retains for future analytical development efforts and comparability testing.
References:
- O'Donnell BT, Ives CJ, Mohiuddin OA, Bunnell BA. Beyond the Present Constraints That Prevent a Wide Spread of Tissue Engineering and Regenerative Medicine Approaches. Front Bioeng Biotechnol. 2019;7:95. Published 2019 May 7. doi:10.3389/fbioe.2019.00095.
- Guidance for Industry and Review Staff. Target Process Profile — A Strategic Development Process Tool, March 2007: https://www.fda.gov/regulatory-information/search-fda-guidance-documents/target-product-profile-strategic-development-process-tool.
- Hollister SJ, Murphy WL. Scaffold translation: barriers between concept and clinic. Tissue Eng Part B Rev. 2011;17(6):459—474. doi:10.1089/ten.TEB.2011.0251.
- Bailey AM, Mendicino M, Au P. An FDA perspective on preclinical development of cell-based regenerative medicine products. Nat Biotechnol. 2014;32(8):721—723. doi:10.1038/nbt.2971.
- Guidance for Industry. Preclinical Assessment of Investigational Cellular and Gene Therapy Products, November 2013: https://www.fda.gov/regulatory-information/search-fda-guidance-documents/preclinical-assessment-investigational-cellular-and-gene-therapy-products.
- Russell WMS and Burch RL. The Principles of Humane Experimental Technique. London: Methuen, 1959.
- Forbes SJ, Gupta S, Dhawan A. Cell therapy for liver disease: From liver transplantation to cell factory. J Hepatol. 2015;62(1 Suppl):S157—S169. doi:10.1016/j.jhep.2015.02.040.
- Jones MK, Lu B, Girman S, Wang S. Cell-based therapeutic strategies for replacement and preservation in retinal degenerative diseases. Prog Retin Eye Res. 2017;58:1—27. doi:10.1016/j.preteyeres.2017.01.004.
- Larochelle A, Dunbar CE. Hematopoietic stem cell gene therapy: assessing the relevance of preclinical models. Semin Hematol. 2013;50(2):101—130. doi:10.1053/j.seminhematol.2013.03.025.
- INTERACT Meetings (Initial Targeted Engagement for Regulatory Advice on CBER products), May 2019: https://www.fda.gov/vaccines-blood-biologics/industry-biologics/interact-meetings-initial-targeted-engagement-regulatory-advice-cber-products.
- Good Laboratory Practice for Nonclinical Laboratory Studies. 21 CFR Part 58, April 2019: https://www.accessdata.fda.gov/scripts/cdrh/cfdocs/cfcfr/CFRSearch.cfm?CFRPart=58.
About The Authors:
 Katy Spink, Ph.D., is COO and managing partner of Dark Horse Consulting. Prior to joining Dark Horse, she was COO of Asterias Biotherapeutics, a publicly traded cell therapy company with clinical stage programs in neurology and immuno-oncology, from 2013 to 2018. Spink previously served as SVP of program operations at Geron Corporation, where she led the cross-functional project teams for all of Geron’s cell therapy programs, including the first human embryonic stem cell product to enter clinical trials. Prior to Geron, Spink was a management consultant at McKinsey & Company. She received her B.A. in biochemistry from Rice University and her Ph.D. in cancer biology from Stanford University as a HHMI Predoctoral Fellow.
Katy Spink, Ph.D., is COO and managing partner of Dark Horse Consulting. Prior to joining Dark Horse, she was COO of Asterias Biotherapeutics, a publicly traded cell therapy company with clinical stage programs in neurology and immuno-oncology, from 2013 to 2018. Spink previously served as SVP of program operations at Geron Corporation, where she led the cross-functional project teams for all of Geron’s cell therapy programs, including the first human embryonic stem cell product to enter clinical trials. Prior to Geron, Spink was a management consultant at McKinsey & Company. She received her B.A. in biochemistry from Rice University and her Ph.D. in cancer biology from Stanford University as a HHMI Predoctoral Fellow.
 Nathan Manley, Ph.D., is a senior consultant at Dark Horse Consulting with over 15 years’ experience in stem cell biology, neuroscience, and preclinical modeling of human disease. Prior to joining Dark Horse, he led research and preclinical development efforts at Asterias Biotherapeutics, a pluripotent stem cell-based biotech company, and trained as an academic researcher in the departments of Biology and Neurosurgery at Stanford University. Over the course of his career, Manley has contributed to the advancement of a wide range of experimental and clinical-stage cell therapy products, authored scientific articles, and co-authored cell therapy patent applications. He also is a part-time instructor in the Biology Department at San Jose State University.
Nathan Manley, Ph.D., is a senior consultant at Dark Horse Consulting with over 15 years’ experience in stem cell biology, neuroscience, and preclinical modeling of human disease. Prior to joining Dark Horse, he led research and preclinical development efforts at Asterias Biotherapeutics, a pluripotent stem cell-based biotech company, and trained as an academic researcher in the departments of Biology and Neurosurgery at Stanford University. Over the course of his career, Manley has contributed to the advancement of a wide range of experimental and clinical-stage cell therapy products, authored scientific articles, and co-authored cell therapy patent applications. He also is a part-time instructor in the Biology Department at San Jose State University.