
A Phase-Appropriate Approach For Assay Validation In Cell & Gene Therapies
By BioPhorum

A phase-appropriate approach to assay validation continues to be a widely accepted and adopted strategy to support the clinical development of general biologics; cell and gene therapies (CGT) are no exception. However, current regulatory guidance is inadequate to sufficiently support the phase-appropriate readiness of analytical assays used in all phases of CGT clinical development and regulatory filing. This has led to a lack of consensus within the cell and gene community on the phase-appropriate development and validation of the analytical assays used at all phases of the CGT product life cycle.
Having insufficient and/or inadequate data packages leads to an increased risk of delays in regulatory filing and approval of the clinical candidates. Given the often-accelerated pace of CGT clinical programs for therapies targeted at patients with unmet medical needs, or where other traditional treatments may have been insufficient, any delays in the regulatory approval process could have a significant impact on the availability of drugs to patients.
This article promotes alignment on a common phase-appropriate approach to analytical assay validation with respect to the critical quality attributes (CQAs) of the most common CGT modalities. The recommendations are aimed at providing a faster and more efficient route to CGT product development that is compliant with regulatory standards.
Product And Methods Life Cycle
CGT products typically aim to treat patients with rare, life-limiting, and/or threatening conditions where no other viable treatment or cure exists. To aid sponsors’ ability to bring these promising therapies to the marketplace as early as possible, regulatory agencies have established industry guidelines and frameworks to expediate the development and approval process.
However, there is insufficient guidance detailing how CGT developers can accelerate chemistry, manufacturing, and controls (CMC) development while ensuring the processes and products are fully characterized and controlled. Also, because CGT modalities are relatively new, many CQAs may not yet be known nor fully understood, and well-defined platform methods for each CQA may not exist.
Each CGT product class has a unique product profile, attribute, manufacturing process, and control requirements. Therefore, each product class requires its own considerations for assay validation. Here, we outline a framework for analytical method life cycle management and provide a phase-appropriate approach for method validation for typical CQAs across the differing CGT modalities.
Figure 1 provides a high-level overview of the CMC efforts generally considered to be important in the drug product development life cycle (blue arrow) relative to a proposed analytical method life cycle approach for CGTs (the green arrow).
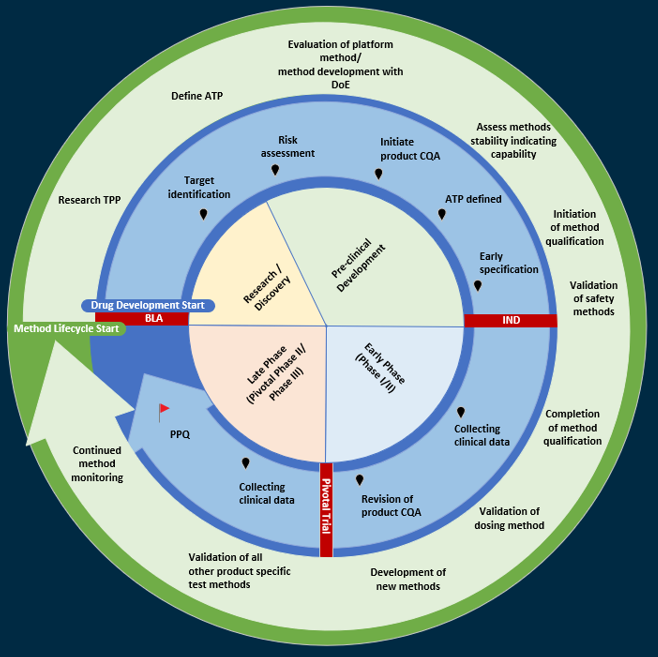
Figure 1: The product and methods life cycle
Key: ATP – Analytical target profiles, BLA – Biologics license application, DoE – Design of experiments, IND – Investigational new drug, PPQ – Process performance qualification, TPP – Target product profiles
The life cycle of analytical methods is closely aligned to the product life cycle. Some of the key steps are:
- Biological products are developed against the target product profile (TPP), which informs the desired attributes of the therapeutic product.
- During preclinical development, product quality risk assessments help inform the potential product CQAs (pCQAs) that, in turn, will inform the phase-appropriate analytical assay validation strategy.
- It is not unusual to have a majority (if not all) of quality attributes considered as pCQAs at preclinical/early clinical development.
- Method development activities are performed to establish methods to address the pCQAs before initiating first-in-human studies, i.e., to meet the established analytical target profile (ATP).
- Method summaries and any available method qualification data are included in the clinical trials application.
- Following a successful clinical trials application filing, the program moves into the early clinical phase to collect data to inform on the safety and efficacy of the therapeutic product.
- CQAs will be refined as new methods are developed/implemented and additional process/product knowledge is gained throughout the clinical development phases.
- As the products move into the pivotal trial phase and process performance qualification lots, method validation studies ensure analytical data is fit for purpose and robust enough to meet performance criteria defined in the ATP.
- Where products have accelerated approval pathways, CMC development timelines may be reduced to support early pivotal trials. Additional agency interactions can be sought to agree on development plans in line with approval timelines.
- Final methods and supporting data are submitted in the biological license application/marketing authorization application filing to move the program into commercial manufacturing.
- Continued method performance monitoring provides ongoing assurance throughout the life cycle.
Analytical methods follow a risk-based method life cycle to ensure they are suitable. Some of the key steps are:
- Throughout preclinical development, analytical method development is initiated and the ATP is defined.
- The ATP is a prospective, technology-independent description of the desired performance of an analytical procedure, and it defines the required quality of the reportable value produced by the procedure.
- The ATP is based on the intended use of the procedure and should include target precision and accuracy (bias), serving as a basis for procedure qualification criteria and a guide for monitoring the procedure during its life cycle. When possible, the target performance should be based on process control strategy requirements.
- Early feasibility assessments may determine the suitability of any preexisting platform methods for a new product; otherwise, a new method(s) may be developed.
- Method development should support the understanding of procedure parameters that may impact assay performances (e.g., sample preparation, number of replicates). Risk assessments are recommended to identify the procedure parameters to investigate.
- Execution of design-of-experiments defines assay parameters for optimum performance and should also evaluate which methods may be capable of assessing product stability.
- As the product moves into early phase clinical development, analytical assay qualification activities are executed to demonstrate that the methods are fit for the intended purpose and perform in line with the ATP.
Analytical Method Bridging
When a new/revised method with improved robustness, sensitivity or accuracy, and operational simplicity is developed to support clinical lot release and stability, replacing the existing method requires a bridging study. Bridging studies may also be required when the previous method is no longer available.
At a minimum, bridging studies should be anchored to a historical, well-established, and qualified or validated method. Typically, it follows on from the new method qualification and/or validation activity.
Based on the International Conference on Harmonisation’s (ICH) Q14 guideline on analytical procedure development (March 2022 draft), the design and extent of the studies needed to support the change include an appropriate bridging strategy to establish the numerical relation between the reportable values of each of the methods and its impact on the specification of the product.
Various approaches can be undertaken, and the selected bridging strategy should be risk-based and depend on the product development stage, ongoing studies, the number and retention of historical batches, etc.
Regulatory Landscape
Reliable analytical methods are required to assure the quality of the therapeutic product with respect to its safety, identity, purity, strength, and/or potency. Sponsors are expected to provide sufficient information in their regulatory submissions relevant to the risks associated with the intended use of the analytical method. A development phase-appropriate approach to generate data regarding the reliability of the method (also known as validation) is allowed by regulatory agencies.
Some guidelines associated with analytical methods development and validation are:
- Chemistry, Manufacturing, and Control (CMC) Information for Human Gene Therapy Investigational New Drug Applications (INDs); Guidance for Industry. FDA, 2020.
- Considerations for the Development of Chimeric Antigen Receptor (CAR) T Cell Products; Draft Guidance for Industry. FDA, 2022.
- Chemistry, Manufacturing, and Control (CMC) Information for Human Somatic Cell Therapy Investigational New Drug Applications (INDs); Guidance for industry. FDA, 2008.
- Potency Tests for Cellular and Gene Therapy Products; Guidance for Industry. FDA, 2011.
- Analytical Procedures and Methods Validation for Drugs & Biologics; Guidance for Industry. FDA, 2014.
- Draft guideline on quality, non-clinical and clinical requirements for investigational advanced therapy medicinal products in clinical trials. EMA, 2018.
- Requirements for quality documentation concerning biological investigational medicinal products in clinical trials. EMA, 2019.
- Guidelines on good manufacturing practice specific for advanced therapies. EMA, 2017.
- Quality, non-clinical and clinical aspects of medicinal products containing genetically modified cells. EMA, 2021.
- Validation of Analytical Procedures. ICH Q2(R2), 2023.
- Analytical Procedure Development. ICH Q14, 2023.
- Analytical Procedure Life Cycle. USP <1220>, 2022.
According to the U.S. FDA’s CMC guidance for investigational gene therapies, validation of analytical procedures is usually not required for original investigational new drug submissions for Phase 1 studies; however, it should be demonstrated that test methods are appropriately controlled. In general, scientifically sound principles for assay performance should be applied. The FDA recommends using compendial methods when appropriate and qualifying safety-related tests before the start of clinical trials. To ensure the safety of gene therapy products, the assays used to determine the dose should be qualified before starting clinical studies. Assays used to measure replication-competent vectors should also meet current FDA recommendations for sensitivity at an early stage of development.
Although the European Medicines Agency has similar requirements, it includes higher expectations for safety tests.
It can be inferred from the above that more information is expected for product safety-related assays early in clinical development. For other assays, there should be sufficient information on suitability, based on their intended use in the manufacturing process.
Determining A Phase-Appropriate Approach To Assay Validation
Pharmaceutical development, as described in ICH Q8(R2), requires that quality attributes are assessed for their criticality to determine their impact on the quality of the final product. The CQAs may be a physical, chemical, biological, or microbiological property, or be characteristics that should be within an appropriate limit, range, or distribution to ensure the desired product quality. CQAs are generally associated with the drug substance, excipients, intermediates (in-process materials), and drug products. Where data are not available, ICH Q11 recognizes that the impact of different product variants and impurities on clinical efficacy, pharmacokinetics, and patient safety can be determined through a risk-based assessment.
As guided by ICH Q8(R2) and ICH Q11, a cascade of interacting elements is defined:
- Create a TPP for the drug being developed.
- Define a quality TPP based on the TPP.
- Establish CQAs using a risk-based assessment.
Typically, product CQAs are generated at the preclinical stage, formally reviewed at each development phase, and finalized before commercialization, thus forming the basis for product specifications and the product and process control strategy. As such, at preclinical and early development stages, CQAs may be considered pCQAs until further data and product knowledge are acquired.
Recommendations For The Scope Of Method Qualification
Analytical methods are required to be validated in accordance with ICH Q2 before a marketing authorization application filing. Although method validation is not required as part of investigational new drug/investigational medicinal product dossier submissions during clinical development, sponsors are required to demonstrate the suitability of the analytical methods for the intended purpose.
There is little regulatory guidance on these requirements beyond ICH Q2(R2), which states: “the scientific principles described in this guideline can be applied in a phase-appropriate manner during clinical development.”
Due to this, the industry has typically applied the term “method qualification” to studies performed to demonstrate the suitability of analytical methods before method validation. However, due to a lack of regulatory guidance, practices may vary between laboratories. See Table 1 for our recommendations for the scope of method qualification.
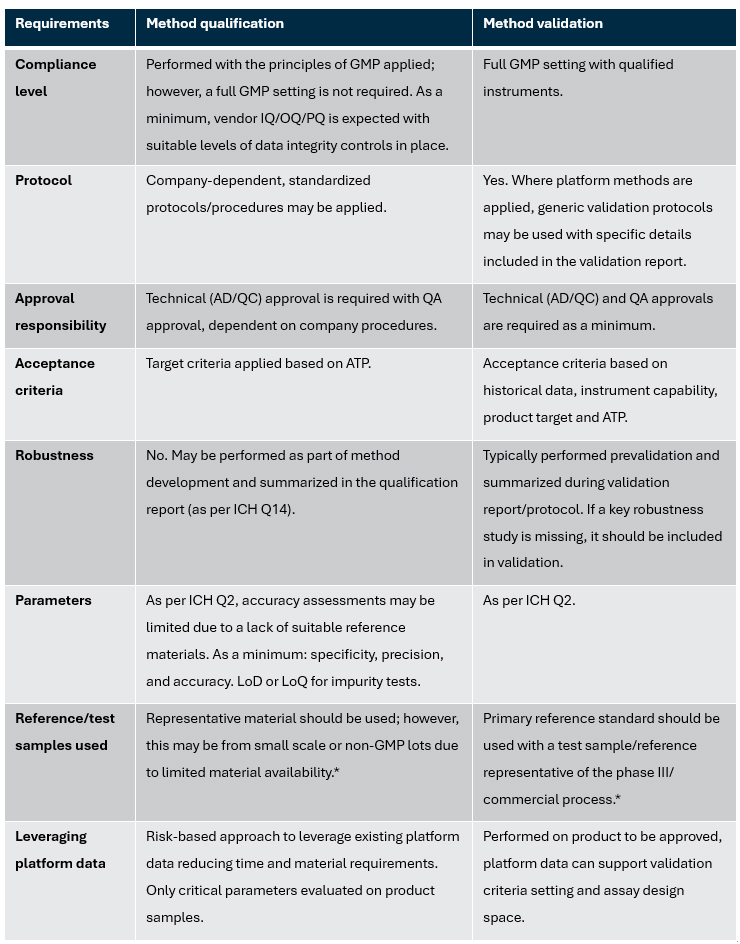
Table 1: Recommendations for scope of method qualification — Comparison of requirements for method qualification and validation
*For stability-indicating methods, degraded samples or spiked impurities should be applied to demonstrate the method as stability-indicating
Key: AD – Analytical development, ATP – Analytical target profiles, GMP – Good manufacturing practice, IQ – Installation qualification, OQ – Operational qualification, LoD – Limit of detection, LoQ – Limit of quantitation, PQ – Performance qualification, QC – Quality control
Future Perspectives
Besides recognizing the often-accelerated pace of CGT clinical programs for therapies that have the potential to be transformative to patients, there are several other aspects to be considered regarding assay validation. One of the common dilemmas is the very limited product yield of CGT programs compared to traditional biological products. Along with the appropriate phase approach to assay validation, minimizing the use of material and meeting the assay validation requirement and intention should be carefully balanced.
The potential use of a platform assay validation approach with product-specific verification (using representative material other than the final products for certain parts of the assay validation or validation without controls) could be considered while developing the assay validation strategy. For potency assays, the appropriate phase approach also can be combined with the matrix approach for overall assay development and validation design.
Unlike traditional biological products, final drug substance and drug product are usually essentially the same material. For CGT products, there are sometimes many critical components used to generate the final drug product. We should also consider the appropriate phase approach of the assay characterization, qualification, or validation strategy associated with those components, as they may not be treated in the same way as traditional drug substance/formulated drug substance.
Along with innovative CGT medicines development, supporting new technologies are also emerging. Some methods that were typically considered as a characterization assay are now implemented more in GxP labs as a release assay. When assessing the technologies, software limitations (especially data integrity) and the method should be evaluated. Selection of the method for the assay release and the phase-appropriate approach for assay validation could be considered together while setting up the product specification.
This article summarizes some of the main points from a recent BioPhorum publication on this topic. To read more, check out the full paper, Industry recommendations for phase appropriate approach for assay validation in cell and gene therapies (CGTs).