
A Guide To Selecting Cell & Gene Therapy Tools, Tech, & Services
By Annie Lamontagne, Blake Bergam, and Uzma Shoukat-Mumtaz, Dark Horse Consulting

The field of cell and gene therapy (CGT) is a diverse group of product classes encompassing pure-play cell therapy, gene-modified cell therapy (both viral and non-viral), pure-play gene therapy, and tissue engineered products. The field has grown rapidly in recent years, expanding from just three FDA approved products in 2016 to 27 licensed products from the Office of Therapeutic Products (OTP) on the market today.1 The speed of development and commercialization in the CGT field is enabled by a strong undercurrent of innovation in supporting tools, technology, and services (TT&S). The landscape of vendors in TT&S market has significantly expanded over the last 10 years and the robust market of today spans multiple value chain segments, including raw materials, process and analytical equipment, software and systems, cold chain and logistics, and service providers. A (non-exhaustive) snapshot of the vendor landscape is shown in Figure 1.
Figure 1: Snapshot of innovative TT&S market landscape across key CGT value chain segments.
CGT developers often aim to establish a baseline manufacturing process in the initial stages of product development, with the intention of optimizing and refining the process throughout the product life cycle. In this article, we provide recommendations to guide the selection process when implementing innovative TT&S into an early-stage manufacturing process that may enable forward compatibility into later stages of development. We’ll focus on a few of the most common decision points across the TT&S value chain segments that we at Dark Horse Consulting encounter.
Process Equipment Selection
The availability of purpose-built, automated technologies designed for CGT processing has significantly increased, with a progression from low-throughput, manual manufacturing scale solutions to those that enable closed-system processing using automated process technology. With so many available options, it can be challenging to navigate the best path forward to select the most suitable equipment alternative. The following steps outline a stepwise approach for a focused evaluation.
Assemble Your Toolbox
In early development the chemistry, manufacturing, and control (CMC) strategy should be built upon current product understanding. Thus, the first step of an equipment selection process should include consultation of your target product profile (TPP) and quality target product profile (QTPP)2 to identify relevant drug product attributes that inform process equipment selection. An illustrative example of how to apply these two key tools to CMC development strategy is provided in Table 1. In the example provided, relevant TPP drug product (DP) attributes inform quality attributes described in the QTPP. Together, the TPP and QTPP attributes guide an assessment of suitability in process equipment decision-making.
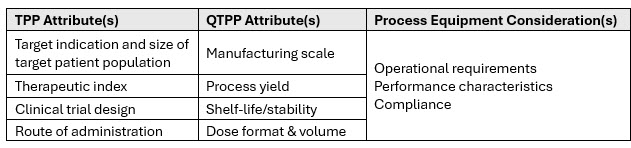
Table 1: Example application of CMC development tools for process equipment selection.
Understand Relevant Regulatory Requirements And Standards
In the U.S., the FDA allows phase-appropriate implementation of the principles of current good manufacturing practices (cGMP). In the U.S. Code of Federal Regulations, 21 CFR Part 211 Subpart D and 21 CFR Part 11 describe requirements for equipment, electronic records, and electronic signatures when used in the manufacture of drug products. In the European Union (EU), requirements are outlined in Eudralex Volume 4, Part 1, Chapter 3. In both examples, the language used is general in nature and applicable to all equipment used in the manufacturing facility.
Agency regulations are wide-ranging, thus a more complete understanding can be attained with a review of industry best practices and relevant standards, such as those detailed in ISO/TS 23565.3 A key difference in the ISO technical specification document is the specialized focus on general requirements for bioprocessing equipment.
Identify Key Attributes And Preferred Features
A distinction between non-product-contacting components and product-contacting components is commonly made with respect to processing equipment. Non-product contacting components include hardware and software, and key attributes may include equipment performance, operational requirements, compliance, and others. Product contacting components primarily encompass single-use consumables in CGT. Example key attributes here include safety, biocompatibility, leachables and extractables, and particulate shedding. It’s important to note that key attributes identified will be process and product specific. For each key attribute, a review of supporting vendor documentation or supplemental sponsor documentation will provide justification that the selected equipment meets minimum requirements or industry best practices.
Additional CMC considerations can be evaluated as program-specific attributes or “preferred features” that may be weighted most heavily in the final selection. These features are key drivers in selection to enable forward compatibility into later stages of development. Example preferred features are shown in Figure 2.
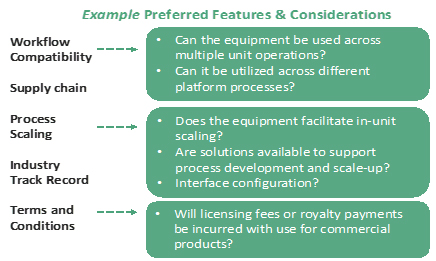
Figure 2: Program-specific attributes that may support later stage CMC development.
Build A Decision Matrix
A decision matrix can aid an objective, comparative assessment of multiple tools and technologies. A two-part assessment incorporating both qualitative and quantitative assessment is recommended. In the qualitative assessment, attributes that are considered a minimum bar for inclusion in the selection process are assigned a binary (yes/no) score to rapidly eliminate unsuitable options. For example, if an equipment option doesn’t readily integrate into existing building management system(s), it may be deprioritized. Overall, the qualitative assessment should narrow the selection to options capable of performing the process as intended and suitable to meet the organization’s strategic goals.
The second component of the decision matrix, qualitative assessment, can be applied to a subset of key attributes using a numerical rating scale. For each attribute included the criteria for conditions that represent “good, better, best” scenarios should be defined. A score is applied to each attribute, and an overall score is calculated across each equipment option being evaluated. Generally, a higher score is considered more favorable. An example quantitative assessment is shown in Figure 3. Additional attributes to consider include technical support, supply chain, scalability, and others.
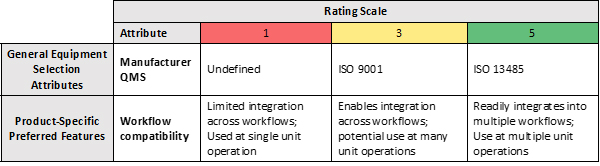
Figure 3: Example Quantitative Assessment Decision Matrix (selected attributes).
The decision matrix described herein is a simple and efficient tool to aid equipment selection, and others can similarly be used. An excellent mini review (Verbarendse et al. 2023) provides an alternative methodology for quantitative evaluation using hypothetical case studies.4
Across a typical CGT workflow there are a bounty of equipment options that support closed-system, automated processing. The recommendations described here provide the framework for a stepwise approach to make more informed and purposeful decisions when implementing innovative process equipment.
Raw Material Selection
Safety is a primary endpoint of first-in-human and early-stage clinical trials; therefore, raw material qualification must be performed as a pre-development activity. Raw material qualification begins with an evaluation of raw material suitability using a risk-based approach. To paraphrase the recent draft FDA draft guidance5,6 on the use of human- and animal-derived materials in CGT manufacturing: The use of human- and animal-derived materials during product manufacturing may increase risks of infectious disease transmission and raises potential safety concerns, such as the possible introduction of adventitious agents or other impurities; thus, these materials should be thoroughly characterized and described in a regulatory submission.
Raw Material Suitability Risk Assessment
A thorough risk management strategy will assess material suitability based on multiple aspects using a methodical approach. A common method for risk assessment of raw material suitability is to perform a failure modes and effects analysis (FMEA). The FMEA identifies critical information for each material and assigns a severity, occurrence, and detectability score, which are multiplied to yield an overall risk priority number. Materials with higher risk priority numbers should have mitigations applied first. Importantly, elements of a raw material suitability risk assessment are often directly included within a regulatory submission.
The following recommendations are helpful when assigning severity, occurrence, and detectability scores and are summarized below in Figure 4.
- Severity: Evaluate the nature of human- and animal-derived materials and use of the material relative to proximity to patient
- Occurrence: Evaluate both the material quality and the manufacturer’s quality management system
- Detectability: Evaluate vendor quality documents to identify gaps in material testing against relevant sourcing, processing, and testing requirements
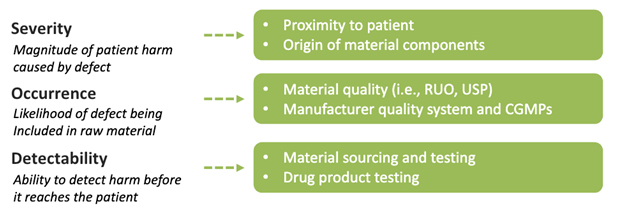
Figure 4: Multiple dimensions of risk in a raw material risk assessment.
Below are two examples when considering severity scoring, specific to proximity to patient:
- Excipients should be injectable quality or tested to a United States Pharmacopoeia (USP) monograph. Novel excipients invite additional scrutiny and will require robust nonclinical data to justify suitability. Changing excipients after stability studies have initiated may also impact the validity of this data.
- Buffers used for reconstitution of media supplements may be reagent quality, include minimal testing, and be sterile filtered prior to use if necessary.
Common Pitfalls And Challenges
Material quality is one of the first indicators of its suitability but is not the whole story. Materials produced under cGMPs or in accordance with ISO certified quality management systems are generally more appropriate than materials intended for research use only (RUO) but in either case it is the sponsor’s responsibility to ensure the materials are appropriately qualified for their specific use. Vendors that advertise materials produced under cGMPs for further manufacturing or for clinical use have likely performed the necessary testing and appropriate sourcing or may be most willing to perform additional mitigations to enhance their products’ suitability. Vendors that advertise RUO materials may not be motivated to assume the burden of creating a version of their product suitable for use in clinical manufacturing as these efforts include high costs and long development timelines.
Understanding a material’s origin and composition is critical to understanding its suitability. One common mistake is misplaced confidence due to marketing messaging. “Xeno-free” materials are not animal-derived but are usually human-derived, which still requires appropriate sourcing, processing, and viral testing. Similarly, “serum-free” and “protein-free” are not regulated terms and do not guarantee an absence of human- or animal-derived components. If a material cannot claim to be produced without the use of human- and animal-derived materials, then a certificate of origin listing each human- and animal-derived component is required.
Raw material offerings for the CGT field are evolving at a rapid rate. The right time to perform a raw material suitability risk assessment is when there is still time to change raw materials or implement necessary mitigations. For unsuitable materials, it may be faster and cheaper to switch to an available alternative cGMP material prior to clinical production than to implement additional safety testing or mitigating process steps after clinical production has begun. Choosing materials based on their performance in the process rather than suitability for patient safety could lead to promising lab data and a stalled clinical program.
Service Provider/CDMO Selection
Selecting a suitable service provider or CDMO partner can be an overwhelming and challenging task. CDMO selection is a lengthy process that requires management of various activities, from collating program requirements to communicating with multiple service providers and evaluating proposals. However, it is possible to streamline the CDMO selection process with the right tools and planning in place.7 An overview of a typical CDMO selection process, informed by the experience from CDMO selections performed by DHC for various types of CGT clients, is shown in Figure 5 and recommendations at a few key steps are provided in the commentary below.

Figure 5: CDMO Selection Process Overview.
Shortlist Your Shortlist
One common mistake is having too long a list of potential CDMO partners when submitting a request for proposal (RFP) or request for quote (RFQ). This can make the evaluation phase complicated and demanding, especially for small teams. It is best to narrow the list down to a manageable size of five or six CDMOs. To help trim the CDMO list, it is important to first define key CDMO attributes important to the success of the program. A few example attributes are mentioned below:
- Location (i.e., U.S. West Coast, Europe, or Asia)
- Process development and optimization capabilities
- Regulatory compliance
- Availability of in-house assays and test methods
- Ability to release drug product material within a designated time frame
Once the potential CDMO list is shortened, CDMOs can be engaged to initiate circulation of non-disclosure agreements (NDAs) for review and signatures. This part of the process takes time, from a few days to a few weeks. So, it best to start communications with CDMOs as early as possible. Here again, the shorter list is advantageous when juggling communications with multiple CDMOs. After the NDAs have been established, introductory calls can be set up to provide a formal introduction between the client and CDMO and discuss a high-level overview of the CDMO capabilities and the client’s program needs.
Establish User Requirements and Specifications
Another common pitfall is not having an established list of user requirements and specifications (URS) for the CGT program prior to engagement with CDMOs. Without a comprehensive URS list, it will be difficult to assess if the CDMO can comply or meet the program needs. URS describes the regulatory and business considerations for process, equipment, facility, operations, personnel, and quality systems. In addition, the CDMO may use the URS to propose strategies to develop and integrate the URS to align with the client’s expectations.
Over-Communicate
From the beginning of engagement with the service providers it is best to set expectations of roles and responsibilities and clearly communicate the planned schedule for the CDMO selection process. This will help keep activities on track and ensure all parties involved are accountable for their tasks. Don’t be worried about reiterating this plan throughout the engagement and ensure all communication is documented. Poor communications can lead to misinterpretation of information, unsatisfactory proposals, and delays in the schedule.
Selecting Lead Candidates
When narrowing the list to top candidates, thoroughly evaluate the proposals. It’s recommended to develop a quantitative matrix to support comprehensive review across each RFP response received. Each CDMO will submit a proposal using their own terminology and fee structure so a system of review that enables an “apples-to-apples” comparison is highly beneficial. Be sure to ask the CDMOs questions to clarify any information. Don’t rush this part of the process! This could lead to selecting a CDMO that may not be able to achieve the program goals. Top candidates should be based on the following criteria:
- Content of proposal aligns with program’s critical considerations
- Acceptable strategy to comply with program’s URS
- Proposed program budget is estimated appropriately according to program requirements and duration
- Proposed timeline aligns with program goals and milestones
Lastly, follow up with on-site visits, quality audits, and a vendor questionnaire to complete the CDMO evaluation phase. Based on a final review of all the compiled information, the most suitable CDMO candidate can then be selected.
Summary
Rapid growth in the CGT field in recent years has been shaped by an upswelling market of supporting tools, technology, and services (TT&S) vendors. While making informed decisions within a crowded landscape can seem daunting, the recommendations provided in this article will support a more structured selection process across the TT&S value chain segments.
References
- https://www.fda.gov/vaccines-blood-biologics/cellular-gene-therapy-products/approved-cellular-and-gene-therapy-products (excludes cord blood derived products and off-market/withdrawn products)
- ICH Harmonised Tripartite Guideline Pharmaceutical Development Q8(R2) (August 2009)
- ISO/TS 23565: Biotechnology — Bioprocessing — General requirements and considerations for equipment systems used in the manufacturing of cells for therapeutic use (2021)
- Verbarendse M, Snyder R, and Lakshmipathy U. Mini-review: Equipment evaluation for process scalability and readiness for current Good Manufacturing Practices in cell therapy workflows. Cytotherapy. 2023 Oct;25(10):1107-1112.
- Draft Guidance for Industry: Considerations for the Use of Human- and Animal-Derived Materials in the Manufacture of Cellular and Gene Therapy and Tissue-Engineered Medical Products (April 2024) https://www.fda.gov/media/178022/download
- Bergam B and Mills S. Understanding FDA's Draft Guidance On Human- And Animal-Derived Materials In The Manufacture Of Cell & Gene Therapy Products. Cell&Gene Guest Column. May 23, 2024.
- Shoukat-Mumtaz U. Streamlining Manufacturing Partner Selection. Unbridled Excellence Webinar Series (August 2023)
 About the Authors:
About the Authors:
Annie Lamontagne, M.S., is a senior consultant at Dark Horse Consulting Group. She has more than 20 years’ experience, including over 10 years exclusively in the CGT field. Lamontagne’s experience ranges across academia and industry with expertise in cell processing, process optimization, clinical manufacturing, technology transfer, and CMC regulatory support.
 Blake Bergam is a senior consultant at Dark Horse Consulting Group with nine years of experience in the field of cell and gene therapy (CGT). He began his CGT career in QA operations, starting up the first internal manufacturing facility at Juno Therapeutics. He then led process and technology development efforts at Juno, including development of a shortened CAR-T manufacturing process and a bespoke cell purification system. Since joining Dark Horse in 2021, Bergam has supported 35+ clients in the areas of CMC strategy, raw material qualification, cGMP operations, and device development.
Blake Bergam is a senior consultant at Dark Horse Consulting Group with nine years of experience in the field of cell and gene therapy (CGT). He began his CGT career in QA operations, starting up the first internal manufacturing facility at Juno Therapeutics. He then led process and technology development efforts at Juno, including development of a shortened CAR-T manufacturing process and a bespoke cell purification system. Since joining Dark Horse in 2021, Bergam has supported 35+ clients in the areas of CMC strategy, raw material qualification, cGMP operations, and device development.
 Uzma Shoukat-Mumtaz is a senior consultant at Dark Horse Consulting Group, with more than 12 years of industry experience in process development, technology transfer, and GMP manufacturing of cell and gene-modified cell therapy products. She has specialized in helping clients with their CDMO selection and, given that range of experience, she builds and expands upon the Dark Horse Consulting CDMO database. Much of her clients’ work also includes either tech transfers or helping clients develop their quality systems.
Uzma Shoukat-Mumtaz is a senior consultant at Dark Horse Consulting Group, with more than 12 years of industry experience in process development, technology transfer, and GMP manufacturing of cell and gene-modified cell therapy products. She has specialized in helping clients with their CDMO selection and, given that range of experience, she builds and expands upon the Dark Horse Consulting CDMO database. Much of her clients’ work also includes either tech transfers or helping clients develop their quality systems.